
Arachidonic acid in Alzheimer's disease
Mélanie H. Thomas1, Sandra Pelleieux1,2, Nicolas Vitale3, Jean Luc Olivier1,2*
Abstract
Alzheimer’s disease is a very complex disease in which neuroinflammation and synaptic dysfunctions play a critical role in association with the two well-known molecular agents of the disease, the Aβ1-42 peptide oligomers and the hyperphosphorylated tau protein. Arachidonic acid, the main member of the ω-6 series, is quantitatively the second polyunsaturated fatty acid in brain and is mainly esterified in membrane phospholipids. It is specifically released by the cytosolic phospholipase A2 whose inhibition or gene suppression counteract the deleterious effects of Aβ1-42 peptide oligomers on cognitive abilities. Arachidonic acid can be reincorporated under the action of the acyl-CoA synthetase 4 and lysophospholipid acyltransferases which remain to be characterized. Free arachidonic acid can be involved in Alzheimer’s disease through several mechanisms. First it is converted by cyclooxygenases-1/2 and the specific prostaglandin synthases into PGE2 and PGD2 which contributes to the occurrence and progression of neuroinflammation. Neuroinflammation has positive as well as negative effects, by favoring Aβ1-42 peptide clearance on one hand and by increasing the production of neurotoxic compounds on the other hand. Second, free arachidonic acid is also involved in synaptic functions as a retrograde messenger and as a regulator of neuromediator exocytosis. Third, some studies indicated that free arachidonic acid and its derivatives activate kinases involved in tau hyperphosphorylation. In addition, the dietary intakes of arachidonic acid in western food increased in the last period. Taken together, these various reports support the hypothesis that arachidonic acid is interesting target in nutrition-based preventive strategies against this disease.
Introduction
All the recent clinical trials against Alzheimer’s disease (AD) failed to evidence any efficiency against the sporadic or late onset cases. A better understanding of AD mechanisms is required to open new therapeutic or preventive strategies. Lipids are important actors in AD as shown by the numerous works devoted to the putative neuroprotective role of docosahexaenoic acid (DHA, C22:6 ω-3), the main polyunsaturated fatty acid (PUFA) in brain1. By contrast, the role of arachidonic acid (ARA, C20:4 ω-6) was less extensively studied, despite the fact that it is the second PUFA in the brain. ARA corresponds to around 20% of the total amount of the neuronal fatty acids and is mainly esterified in membrane phospholipids. After its release by phospholipases A2, ARA is converted by several enzymes into numerous eicosanoids which are actors of neuroinflammation2. But ARA can also be directly involved in synaptic functions as a free fatty acid3. Therefore, the level of intracellular free ARA and the balance between the releasing enzymes and those which allows its incorporation in membrane phospholipids can be critical for AD-associated phenomena such as neuroinflammation and synaptic dysfunctions. Both these events can be observed in AD murine models before the two historical pathognomonic AD signs, the amyloid plaques and the neurofibrillary tangles which are respectively formed by the two known AD agents, Aβ peptide and hyperphosphorylated tau. Finally, western food which contains excessive ω-6 / ω-3 ratios4 (above the recommended value of 5) might favor its incorporation in brain lipids and its influence on AD mechanisms. As a consequence, a better knowledge of the relationship between ARA and these mechanisms is useful for the therapy or prevention of AD. These different points will be discussed in this mini-review.
Mobilization of arachidonic acid in neuronal and glial cells
Free ARA is released from membrane phospholipids by several phospholipases A2 which constitute a superfamily including intracellular as well as secretory and Ca2+-dependent or -independent enzymes5. Group IVA PLA2 (or cPLA2-α) is the only PLA2which specifically release ARA6,7. This cytosolic enzyme is activated and translocated to membranes after a rise of cytosolic calcium concentration and the phosphorylation of its Ser505 by the MAP-kinase cascade7 (Figure 1, left). The involvement of cPLA2–α in AD was first evidenced by its higher expression in AD patient brain8. Several works also showed that cPLA-α mediates the effects of Aβ1-42 peptide in astrocytes and microglial cells9,10. cPLA2-α is also expressed in neuronal cells11,12 which are protected from the pro-apoptotic effects of Aβ1-42 peptide oligomers by the reduction of its activity or expression11. Aβ1-42peptide oligomers activate cPLA2–α through the stimulation of MAP kinase cascade (Figure 1, left) since Erk1/2 inhibitors suppress the cPLA2–α phosphorylation. Several signaling pathways downstream of cPLA2-α activation have been characterized in neuronal cells such as stimulation of sphingomyelinase13 or PLD214 activities. Finally, the critical role of cPLA2-α was definitively established by in vivo experiments showing that its gene suppression leads to resistance to Aβ peptide in a transgenic murine line bearing a double mutation of human APP12 or in wild-type mice submitted to intracerebroventricular injections of Aβ peptide15. But the way by which cPLA2-α-released ARA mediates Aβ1-42 oligomer neurotoxicity remains to be identified.
Free ARA is incorporated into membrane phospholipids through two steps, 1) its esterification with Coenzyme A into arachidonyl-CoA by acyl-CoA synthetases (ACSLs), 2) its transfer from arachidonyl-CoA to lysophospholipids by various acyl-CoA lysophospholipids acyltransferases (LPATs). Among the various ACSLs, ACSL4 specifically esterifies ARA (Figure 1, left) and was first described in steroidogenic tissues16. In addition to the ubiquitous isoform, a neuronal specific isoform was identified and differs from the first one by an additional 41 amino-acid N-terminal domain17. Mutant ACLS4 gene was associated to X-linked mental retardation18 and has been involved in neuronal differentiation, axonal transportations and synapse formation17,18,19. Channeling of ARA by ACSL4 and Lysophosphatidylinositol-acyltransferase-1 (LPIAT1) to phosphatitylinositol (PI) has been described in CHO cells20. But the role of the various LPATs in the ARA incorporation in brain phospholipids remains to be characterized in more details. ACLS4 and LPATs could reduce the free ARA level in neuronal and glial cells, thus limiting the impact of enhanced cPLA2–α activity on this level and the consequences on neuroinflammation and synapse dysfunctions.
Neuroinflammation and AD: role of arachidonic acid and its derivatives
Neuroinflammation plays a positive role in the early AD steps by contributing to Aβ peptide clearance but can contribute to synapse alterations in the advanced stage of the disease when Aβ peptide accumulates and aggregates in amyloid plaques21. Eicosanoids formed from free ARA are key mediators in neuroinflammation. Among the various enzymes which produced these eicosanoids, cyclooxygenases-1 and -2 (COX-1/COX-2) convert ARA into PGH222. Then, PGH2 is used by various prostaglandin synthases to produce the various prostaglandins in the different tissues23. Nonsteroidal anti-inflammatory drugs (NSAIDs ) are well known inhibitors of COX-1 and COX-2. Their involvement in AD has first been suggested by studies which evidenced a reduction of AD risk in patients treated by NSAIDS24-26. In the majority of organs and tissues, COX-1 is constitutively expressed and COX-2 gene expression is induced especially in case of inflammation. On the contrary, brain tissues and especially neuronal cells constitutively express COX-2 in basic condition27 (Figure 1, left). But COX-2 overexpression in brain of AD patients has been reported and correlated to the progression of the disease in several studies28,29. Since Aβ1-42 oligomers do not induce the COX-2 expression in human microglial cell primary cultures30, the COX-2 overexpression observed in AD patient brains is likely to be generated in neuronal cells. On the contrary to COX-2, COX-1 is overexpressed by microglial cells around amyloid plaques31. In addition, the expression of COX-3, a splice variant of COX-1, has also been reported in AD hippocampus and Aβ1-42 treated neuronal primary culture neuronal cells32. PGH2 can be secreted by neuronal cells and converted into other prostaglandins by glial cells. In the brain, the most abundant prostaglandins are PGD2 and PGE2 (Figure 1, right), which are synthetized by several PGD2 and PGE2 synthases. Expression levels of hematopoietic PGD2 synthase (one of the two PGD2 synthases expressed in the brain) are enhanced in microglial cells and astrocytes of AD transgenic mice and patients33. As a result PGD2 is overproduced in these glial cells surrounding the amyloid plaques33. PGE2 binds to several receptors and have opposite effects on inflammation depending on the receptor type. EP2 receptor mediates negative inflammatory effects (Figure 1, right) since its gene suppression reduces the microglial toxic inflammatory response and restored Aβ clearance34. By contrast, suppression of the EP4 receptor increases the inflammatory response in the brain of the APPSwe-PS1ΔE9 mouse35.
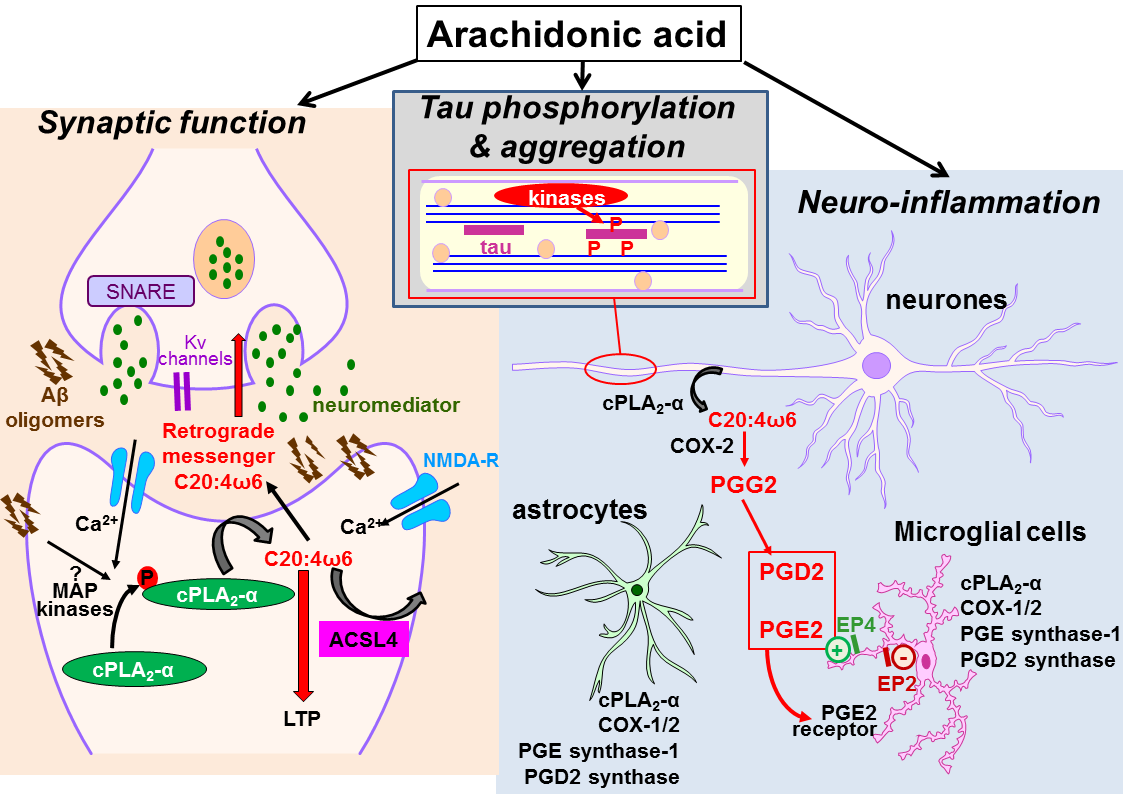
Figure 1: Pleiotropic effects of arachidonic acid in Alzheimer’s disease:
Left: arachidonic acid (ARA) is released by cytosolic phospholipase A2 (cPLA2-α) in neuronal cells and glial cells. It is re-incorporated in phospholipids after its conversion into arachidonyl-CoA by acyl-CoA synthetase 4 (ACSL4). Free ARA influences synaptic function by acting as retrograde messenger and by opening Kv channels which lead to exocytosis of neuromediator and induction of LTP.
Top: arachidonic acid activates several kinases which directly or indirectly contribute to increase tau phosphorylation levels.
Right: free Arachidonic acid is converted into PGG2 by cyclooxygenase-2 (COX-2) which is overexpressed in neuronal cells in Alzheimer’s disease. PGG2 is converted in PGD2 and PGE2 by corresponding prostaglandin synthases in astrocytes and microglial cells. PGE2 bind to EP2 and EP4 receptors on microglial cell membranes. These receptors have different effects in neuroinfllammation, stimulation of Aβ clearance or production of neurotoxic molecules. These positive or negative effects illustrate the dual role of neuroinflammation.
Five-lipoxygenase (5-LOX) converts free ARA into 5-hydroxyperoxyeicosatetraenoic acid [5-HPETE], which is then stabilized into 5-hydroxyeicosatetraenoic acid (5-HETE) or converted by 5-LOX into leukotriene A4 (LTA4). 5-LOX gene deletion attenuates the worsening effect of LPS-induced neuroinflammation in transgenic AD-model mice36. But the use of 5-LOX inhibitors demonstrated that 5-LOX and its products have a stronger impact on Aβ peptide production and tau phosphorylation in neuronal cells37 than in the proliferation of glial cells and neuroinflammation.
Despite the data of retrospective epidemiologic studies which reported a lower AD risk in NSAIDS-treated populations, randomized controlled trial failed to support the efficiency of these drugs in the AD therapy26,38. Therefore, other approaches are needed to explain the discrepancy between the data of the observational studies or the works on AD animal models on one hand and those of the clinical trials on the other hand. Parallel mechanisms could explain the role of COX inhibitors in AD. For example, higher plasma concentrations of tryptophan metabolites were found in AD patients suggesting the involvement of the kynurenine pathway which influences the serotoninergic and glutamatergic neurotransmission39. Ibuprofen drastically reduced the expression of the neuronal tryptophan 2,3-dioxygenase (Tdo2), which encodes an enzyme that metabolizes tryptophan to kynurenine while Tdo2 inhibition prevented behavioural deficits in the AD model APPSwe-PS1ΔE9 mice40. However, the influence of ARA and its metabolites on Tdo2 expression remains to be established.
Arachidonic acid and synaptic functions
Before its deposit in amyloid plaques, Aβ1-42 peptide forms oligomers which are now recognized as the main pathological actor in the early steps of AD. These oligomers initially alter the remodeling of the synaptic network and memory abilities by inhibiting long-term potentiation (LTP)41. Free ARA produced by the postsynaptic neuron acts as a retrograde messenger which increases the neurotransmitter release by the presynaptic neuron and results in the induction of LTP42 (Figure 1, left). Regulation of the pre and post-synaptic Kv channels by ARA is also involved in its effect on LTP43. Moreover the LTP was reduced by harmonize cPLA2-alpha gene suppression or its inhibition by arachydonyl trifluoromethyl ketone (AACOCF3)44. Thus ARA seems to be necessary for the LTP induction.
Free ARA activates the soluble N ethylmaleimide-attachment receptors (SNARE), which are required for the fusion of synaptic vesicles with the plasma membrane45 (Figure 1, left). Vesicle SNARE protein VAMP-2 interacts with the plasma membrane SNARE proteins SNAP25 and syntaxin-1 to form a complex which promotes membrane fusion. ARA induces the binding of syntaxin-1 to the SNARE complex in presence of Munc18-1, which is a critical regulator of this process46. The involvement of ARA in the vesicle membrane fusion was also demonstrated by the formation of TIP30 complex which associates Tip30 endophilin B and ACSL447. In this complex, TIP30 transfers ACSL4-produced arachidonyl-CoA onto phosphatidic acid and thus forms new species which induce close contact between membranes. Initial work on neuroendocrine cells revealed increased ARA production during exocytosis and clearly showed that ARA provision facilitated vesicular release48. Taken together, these studies support the hypothesis that modulation of free ARA levels can impact synaptic functions. Excessive maintenance of LTP or release of neurotransmitter could result from excessive levels of free ARA levels and have detrimental effects on memory storage.
Arachidonic acid and tau
Tau is a microtubule associated protein which is physiologically weakly phosphorylated and concentrated in the neuronal axons49(Figure, top). The C-terminal domain of Tau binds to α and β-tubulin to assemble microtubules and regulates the axonal transport of neurotransmitters. In AD, an excessive kinase activity and/or a shortfall of phosphatase activity lead to Tau hyperphosphorylation49. Hyperphosphorylated Tau accumulates in the dendrites and cell body of the neuron, forms helical filaments and subsequently neurofibrillary tangles.50
Several studies reported that free ARA in vitro induced tau polymerization. The ARA concentration required for the polymerization differs according to the various tau isoforms which are generated by alternative splicing51. These polymers activate microglial cells52.
Among the various kinases which phosphorylate tau, two enzymes, PKNα and PKCξ bind and are activated by ARA53,54. PKNα is one of the 3 PKC-homologous serine/protein kinases (PKN), is highly expressed in brain54 and accumulates in neurofibrillary tangles55,56. Increased expression levels of PKCξ, an atypical Protein Kinase C family member, have been measured in neuronal membrane fraction of AD TG2576 mice57 and in T-lymphocytes of patients affected by severe forms of AD58. PKCξ contributes to tau phosphorylation by targeting leucine-rich repeat kinase 2 [LRRK2] whose mutation is the most frequent cause of the genetic forms of Parkinson disease59.
Lipoxygenase-generated derivatives of ARA are also involved in tau phosphorylation. Suppression of 5-LOX gene expression improves Tau and Aβ-related mechanisms in AD mouse models60. In addition, the dual enzyme 12/15-lipoxygenase and its products, 12(S)-HETE and 15(S)-HETE modulates Tau metabolism specifically via the cdk5 kinase pathway61.
Conclusion
Free ARA contributes to AD progression through various mechanisms. Through its conversion into pro-inflammatory eicosanoids, it participates to neuroinflammation. ARA is directly involved in synaptic functions as a retrograde messenger and a regulator of neuromediator exocytosis. Finally, some works also indicate that ARA might have some influence on tau phosphorylation and polymerization. All these data shows that ARA has pleiotropic effects in AD and might be an interesting target in the fight against this complex disease.
ARA is provided by diet, directly from animal products and indirectly from the conversion of linoleic acid found in the vegetal oils. Linoleic acid and ARA amounts increased in the last 40 years period in the western diets. Therefore the influence of dietary ARA on the occurrence of AD is an important issue for the prevention of the disease. Unfortunately, only two studies were performed on transgenic AD murine model to measure the impact of dietary ARA on the pathological process and found opposite results on Aβ production and deposition62,63. Therefore, additional studies are necessary to clarify the dietary ARA impact in AD and to identify the underlying mechanisms, keeping in mind the perspective of nutrition-based preventive strategies of AD.
Acknowledgements
We thank the French France-Alzheimer association and the French region of Lorraine for the financial support to our work on the role of dietary arachidonic acid in Alzheimer’s disease which was the initiation point of this minireview.
References
- Luchtman D. W. and Song C. Cognitive enhancement by omega-3 fatty acids from child-hood to old age findings from animal and clinical studies. Neuropharmacology. 2013; 64: 550–565.
- Funk C. D. Prostaglandins and leukotrienes advances in eicosanoid biology. Science. 2001; 294(5548): 1871–1875.
- Latham C F, Osborne S L, Cryle M J et al. Arachidonic acid potentiates exocytosis and allows neuronal SNARE complex to interact with Munc18a. J Neurochem. 2007; 100(6): 1543–1554.
- Rett B S, Whelan J. Increasing dietary linoleic acid does not increase tissue arachidonic acid content in adults consuming Western type diets a systematic review. Nutr Metab. 2011; 8: 36.
- Burke JE, Dennis EA. Phospholipase A2 structure/function mechanism and signaling. J Lipid Res. 2009; 50 Suppl: S237-42.
- Clark JD, Lin LL, Kriz RW, et al. A novel arachidonic acid-selective cytosolic PLA2 contains a Ca(2+)-dependent translocation domain with homology to PKC and GAP. Cell. 1991; 65(6): 1043-51.
- Leslie CC. Cytosolic phospholipase A?: physiological function and role in disease. J Lipid Res. 2015; 56(8): 1386-402.
- Stephenson DT, Lemere CA, Selkoe, DJ, et al. Cytosolic phospholipase A2 (cPLA2) immunoreactivity is elevated in Alzheimer's disease brain. Neurobiol Dis. 1996; 3(1): 51-63.
- Hicks JB, Lai Y, Sheng W, et at al. Amyloid-beta peptide induces temporal membrane biphasic changes in astrocytes through cytosolic phospholipase A2. Biochim Biophys Acta. 2008; 1778(11): 2512-9.
- Szaingurten-Solodkin I, Hadad N, Levy R. Regulatory role of cytosolic phospholipase A2alpha in NADPH oxidase activity and in inducible nitric oxide synthase induction by aggregated Abeta1-42 in microglia. Glia. 2009; 57(16): 1727-40.
- Kriem B, Sponne I, Fifre A, et al. Cytosolic phospholipase A2 mediates neuronal apoptosis induced by soluble oligomers of the amyloid-beta peptide. FASEB J. 2005; 19(1): 85-7.
- Sanchez-Mejia RO, Newman JW, Toh S, et al. Phospholipase A2 reduction ameliorates cognitive deficits in a mouse model of Alzheimer's disease. Nat Neurosci. 2008; 11(11): 1311-8.
- Malaplate-Armand C, Florent-Béchard S, Youssef I, et al. Soluble oligomers of amyloid-beta peptide induce neuronal apoptosis by activating a cPLA2-dependent sphingomyelinase-ceramide pathway. Neurobiol Dis. 2006; 23(1): 178-89.
- Oliveira TG, Chan RB, Tian H, et al. Phospholipase d2 ablation ameliorates Alzheimer’s disease-linked synaptic dysfunction and cognitive deficits. J Neurosci Off J Soc Neurosci. 2010; 30(49): 16419 28.
- Desbène C, Malaplate-Armand C, Youssef I, et al. Critical role of cPLA2 in Aβ oligomer-induced neurodegeneration and memory deficit. Neurobiol Aging. 2012; 33: 1123 e17-29.
- Kang MJ, Fujino T, Sasano H, et al. A novel arachidonate-preferring acyl-CoA synthetase is present in steroidogenic cells of the rat adrenal; ovary; and testis. Proc Natl Acad Sci U S A. 1997; 94(7): 2880-4.
- Cho, YY. A novel role of brain-type ACS4 isotype in neuronal differentiation. Biochem Biophys Res Commun. 2012 ; 419(3): 505-10.
- Meloni I, Parri V, De Filippis R, et al. The XLMR gene ACSL4 plays a role in dendritic spine architecture. Neuroscience. 2009; 159(2): 657-69.
- Liu Z, Huang Y, Zhang Y, et al. Drosophila Acyl-CoA synthetase long-chain family member 4 regulates axonal transport of synaptic vesicles and is required for synaptic development and transmission. J Neurosci. 2011; 31(6): 2052-63.
- Küch EM, Vellaramkalayil R, Zhang I, et al. Differentially localized acyl-CoA synthetase 4 isoenzymes mediate the metabolic channeling of fatty acids towards phosphatidylinositol. Biochim Biophys Acta. 2013; 1841(2): 227-239.
- Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015; 14: 388-405
- Smith WL, Song I. The enzymology of prostaglandin endoperoxide H synthases 1 and 2. Prostaglandins Other Lipid Mediat. 2002; 68 69: 115 28.
- Pecchi E, Dallaporta M, Jean A, et al. Prostaglandins and sickness behavior old story new insights. Physiol Behav. 2009; 97(3 4): 279 92.
- Szekely CA, Zandi PP. Non-steroidal anti-inflammatory drugs and Alzheimer's disease the epidemiological evidence. CNS Neurol Disord Drug Targets. 2010; 9(2): 132-9.
- Côté S, Carmichael PH, Verreault R, et al. Nonsteroidal anti-inflammatory drug use and the risk of cognitive impairment and Alzheimer's disease. Alzheimers Dement. 2012 May; 8(3): 219-26.
- Wang J, Tan L, Wang HF, et al. Anti-inflammatory drugs and risk of Alzheimer's disease, an updated systematic review and meta-analysis. J Alzheimers Dis. 2015; 44(2): 385-96.
- Minghetti, L. Cyclooxygenase-2 [COX-2] in inflammatory and degenerative brain diseases. J Neuropathol Exp Neurol. 2004; 63(9): 901-10.
- Ho L, Purohit D, Haroutunian V, et al. Neuronal cyclooxygenase 2 expression in the hippocampal formation as a function of the clinical progression of Alzheimer disease. Arch Neurol. 2001; 58(3): 487 92.
- Fujimi K, Noda K, Sasaki K, et al. Altered expression of COX-2 in subdivisions of the hippocampus during aging and in Alzheimer’s disease the Hisayama Study. Dement Geriatr Cogn Disord. 2007; 23(6): 423 31.
- Hoozemans JJ, Veerhuis R, Janssen I, et al. The role of cyclo-oxygenase 1 and 2 activity in prostaglandin E secretion by cultured human adult microglia, implications for Alzheimer's disease. Brain Res. 2002; 951(2): 218-26.
- Yermakova AV, Rollins J, Callahan LM, et al. Cyclooxygenase-1 in human Alzheimer and control brain quantitative analysis of expression by microglia and CA3 hippocampal neurons. J Neuropathol Exp Neurol. 1999; 58(11): 1135-46.
- Cui JG, Kuroda H, Chandrasekharan NV, et al. Cyclooxygenase-3 gene expression in Alzheimer hippocampus and in stressed human neural cells. Neurochem Res. 2004; 29(9): 1731-7.
- Mohri I, Kadoyama K, Kanekiyo T, et al. Hematopoietic prostaglandin D synthase and DP1 receptor are selectively upregulated in microglia and astrocytes within senile plaques from human patients and in a mouse model of Alzheimer disease. J Neuropathol Exp Neurol. 2007; 66(6): 469 80.
- Johansson JU, Woodling NS, Wang Q, et al. Prostaglandin signaling suppresses beneficial microglial function in Alzheimer’s disease models. J Clin Invest. 2015; 125(1): 350 64.
- Woodling NS, Wang Q, Priyam PG, et al. Suppression of Alzheimer-associated inflammation by microglial prostaglandin-E2 EP4 receptor signaling. J Neurosci Off J Soc Neurosci. 2014; 34(17): 5882 94.
- Joshi YB, Giannopoulos PF, Chu J, et al. Modulation of lipopolysaccharide-induced memory insult γ-secretase and neuroinflammation in triple transgenic mice by 5-lipoxygenase. Neurobiol Aging. 2014; 35(5): 1024-31.
- Chu J, Li JG, Praticò D. Zileuton improves memory deficits amyloid and tau pathology in a mouse model of Alzheimer's disease with plaques and tangles. PLOS One. 2013; 8: e70991.
- Miguel- Álvarez M1, Santos-Lozano A, Sanchis-Gomar F, et al. Non-steroidal anti-inflammatory drugs as a treatment for Alzheimer's disease a systematic review and meta-analysis of treatment effect. Drugs Aging. 2015; 32(2): 139-47.
- Gulaj E, Pawlak K, Bien B, et al. Kynurenine and its metabolites in Alzheimer's disease patients. Adv Med Sci. 2010; 55(2): 204-11.
- Woodling NS, Colas D, Wang Q, et al. Cyclooxygenase inhibition targets neurons to prevent early behavioural decline in Alzheimer's disease model mice. Brain. 2016; 139(Pt 7): 2063-81.
- Shankar GM, Li S, Mehta TH, et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med. 2008; 14(8): 837 42.
- Nishizaki T, Nomura T, Matsuoka T, et al. Arachidonic acid as a messenger for the expression of long-term potentiation. Biochem Biophys Res Commun. 1999; 254(2): 446 9.
- Angelova PR, Müller WS. Arachidonic acid potently inhibits both postsynaptic-type Kv4.2 and presynaptic-type Kv1.4 IA potassium channels. Eur J Neurosci. 2009; 29(10): 1943 50.
- Su LD, Wang DJ, Yang D, et al. Retrograde cPLA2α/arachidonic acid/2-AG signaling is essential for cerebellar depolarization-induced suppression of excitation and long-term potentiation. Cerebellum. 2013; 12(3): 297-9.
- Jahn R, Scheller RH. SNAREs--engines for membrane fusion. Nat Rev Mol Cell Biol. 2006; 7(9): 631 43.
- Latham CF, Osborne SL, Cryle MJ, et al. Arachidonic acid potentiates exocytosis and allows neuronal SNARE complex to interact with Munc18a. J Neurochem. 2007; 100(6): 1543 54.
- Zhang C, Li A, Gao S, et al. The TIP30 protein complex, arachidonic acid and coenzyme A are required for vesicle membrane fusion. PloS One. 2011; 6(6): e21233.
- Vitale N, Thiersé D, Bader MF. Melittin promotes exocytosis in neuroendocrine cells through the activation of phospholipase A?. Regul Pept. 2010; 165(1): 111 6.
- Khan SS, Bloom GS. Tau The Center of a Signaling Nexus in Alzheimer'sDisease. Front Neurosci. 2016; 9: 10 31.
- Billingsley ML, Kincaid RL. Regulated phosphorylation and dephosphorylation of tau protein effects on microtubule interaction intracellular trafficking and neurodegeneration. Biochem J. 1997; 323(Pt 3): 577–591.
- King ME, Gamblin TC, Kuret J, et al. Differential assembly of human tau isoforms in the presence of arachidonic acid. J Neurochem. 2000; 74(4): 1749-57.
- Morales I, Jiménez JM, Mancilla M, et al. Tau oligomers and fibrils induce activation of microglial cells. J Alzheimers Dis. 2013; 37(4): 849-56.
- Kochs G, Hummel R, Meyer D et al. Activation and substrate specificity of the human protein kinase C alpha and zeta isoenzymes. Eur J Biochem. 1993; 216: 597-606.
- Mukai, H. The structure and function of PKN a protein kinase having a catalytic domain homologous to that of PKC. J Biochem. 2003; 133: 17-27.
- Kawamata T, Taniguchi T, Mukai H, et al. A protein kinase PKN accumulates in Alzheimer neurofibrillary tangles and associated endoplasmic reticulum derived vesicles and phosphorylates tau protein. J Neurosci. 1998; 18(18): 7402-10.
- Taniguchi T, Kawamata T, Mukai H, et al. Phosphorylation of tau is regulated by PKN. J Biol Chem. 2001; 276(13): 10025-31.
- Rossner S, Mehlhorn G, Schliebs R, et al. Increased neuronal and glial expression of protein kinase C isoforms in neocortex of transgenic Tg2576 mice with amyloid pathology. Eur J Neurosci. 2001; 13(2): 269-78.
- Miscia S, Ciccocioppo F, Lanuti P, et al. Abeta(1-42) stimulated T cells express P-PKC-delta and P-PKC-zeta in Alzheimer disease. Neurobiol Aging. 2009; 30(3): 394-406.
- Zach S, Felk S, Gillardon F. Signal transduction protein array analysis links LRRK2 to Ste20 kinases and PKC zeta that modulate neuronal plasticity. PLoS One. 2010; 5: e13191.
- Joshi YB, Giannopoulos PF, Chu J, et al. Absence of ALOX5 gene prevents stress-induced memory deficits synaptic dysfunction and tauopathy in a mouse model of Alzheimer’s disease. Hum Mol Genet. 2014; 23(25): 6894 902.
- Giannopoulos PF, Joshi YB, Chu J, et al. The 12-15-lipoxygenase is a modulator of Alzheimer’s-related tau pathology in vivo. Aging Cell 2013; 12(6): 1082 90.
- Hosono T, Nishitsuji K, Nakamura T, et al. Arachidonic acid diet attenuates brain Aβ deposition in Tg2576 mice. Brain Res. 2015; 1613: 92-9.
- Amtul Z, Uhrig M, Wang L, et al. Detrimental effects of arachidonic acid and its metabolites in cellular and mouse models of Alzheimer’s disease, structural insight. Neurobiol Aging. 2012; 33(4): 831 e21-31.