
Enhancement of Local Retionic Acid Signaling: A Pivotal Mechanism in Fluoxetine's Pleiotropic Actions
Vera Clemens, Francesca Regen, Julian Hellmann-Regen*
Abstract
Major depression (MDD) is one of the leading global causes of all non-fatal burden of disease. Involving monoaminergic imbalances, but also hormonal, structural and inflammatory alterations, the underlying pathogenesis remains incompletely understood. The antidepressant drug fluoxetine, which may be considered the “prototype” of all selective serotonin reuptake inhibitors (SSRI), appears to affect all of these processes. Interestingly, this is also the case for retinoic acid (RA), the highly potent active metabolite of vitamin A. In this review, we discuss RA signaling as a central mechanism of action – and missing link – for the multiple, pleiotropic effects of fluoxetine in the CNS, suggesting that direct inhibition of CYP-450-mediated RA catabolism by fluoxetine results in increased local concentration, and enhanced paracrine RA signaling in the CNS.
Introduction
Mental and substance use disorders are the leading global cause of all non-fatal burden of disease with major depressive disorder (MDD) having the biggest impact in this group1. While the neurobiological mechanisms underlying MDD are highly complex and not yet fully understood, treatment with antidepressant substances, first serendipity discovered, started over 50 years ago. Revelation of a common monoaminergic mode of action has significantly contributed to the neurobiological understanding of MDD pathogenesis and led to the “monoamine hypothesis”2. Fluoxetine, which was the first selective serotonin reuptake inhibitor (SSRI) that was approved by the Food and Drug Administration (FDA) in 1987, may be considered the “prototype” of all modern, selective monoaminergic antidepressants. Although numerous substances with a selective monoaminergic mechanism of action have followed and been approved since, fluoxetine is still effectively used in clinical practice. Fluoxetine, but also other selective monoaminergic antidepressants, do not exhibit any immediate antidepressant actions. In fact, antidepressant action is known to occur no earlier than 2-3 weeks after onset of treatment, a fact suggesting other processes, secondary to (altered) monoaminergic signaling, to be responsible for and involved in an antidepressant mechanism of action. Therefore, many additional theories about fluoxetine’s antidepressant mode of action are discussed. Several specific, but likely “serotonin-independent” effects of fluoxetine have been demonstrated, including enhancement of synaptic plasticity in hippocampal neurons3,4 anti-inflammatory mechanisms1, but also a serotonin-independent effect on the acid sphingomyelinase-ceramide system6,7.
Interestingly, one of the hallmark characteristics of fluoxetine is its rather long half-life and its ability to interact strongly with the cytochrome P450 (CYP450)-system30. While this fact has mainly been of interest in the context of drug-drug interactions, it is noteworthy that CYP450 enzymes are rather ubiquitously expressed, also (or especially) in the brain, where they are believed to contribute to specific local small molecule levels.
The metabolism of retinoids
Retinoic acid (RA), the active metabolite of Vitamin A, is one of the most important, CNS-active small molecules regulated by local CYP450-catabolism. As mentioned above, fluoxetine is known to inhibit a number of likely RA-degrading CYP450-isozymes30. Therefore, a local interaction in the brain may be of relevance for local RA concentrations as well. Most interestingly, we were able to demonstrate this relationship in a series of previous experiments, indicating a direct inhibitory effect of fluoxetine on local RA degradation in brain tissue8, pointing towards altered RA-signaling as a putative missing link between the multiple, RA-like (and serotonin-independent) effects of fluoxetine. RA is a crucial CNS morphogen, involved in patterning and neuronal differentiation in embryonic brain development, but also an essential factor in neuronal plasticity and regeneration of the adult CNS9. RA belongs to the group of endogenous retinoids, which are metabolites of retinol and other precursors termed “vitamin A” or beta-carotene. After oral uptake, vitamin A-derivatives are stored as retinyl esters, mainly in the liver10. Transport to tissue targets takes place via the blood as retinol bound to retinol-binding protein (RBP)11. Uptake into the target cells has been demonstrated to involve binding to membrane receptor STRA612. Subsequently, retinol is oxidized by the retinol dehydrogenase (ROLDH) into retinal and in a second step via retinaldehyde dehydrogenase (RALDH) into RA, the final active metabolite which can diffuse to other, neighboring cells, generating “RA gradients”, which again depend not only on the local rate of synthesis, but more importantly on the local rates of degradation via the CYP450-system. Resembling a central, endogenous neurotrophic process, an involvement of altered RALDH function has been suggested by Gruenblatt and Riederer to play a role in the pathogenesis of neurodegenerative disorders such as Alzheimers‘ disease at the genetic, protein expression level and at the level of enzymatic activity13. With respect to RA’s molecular actions, after binding to cellular retinoic acid binding protein (CRABP) I and II, CRABP II assists RA to enter the nucleus and to reach its nuclear receptors14. RA binds to retinoic acid receptors (RARα, RARβ and RARγ) and retinoic X receptors (RXRα, RXRβ and RXRγ) which in turn bind to retinoic acid response elements (RARE), inducing ligand-dependent transcription15.
RA signaling in MDD
RA is involved in several MDD related processes, which is most strikingly demonstrated in a knockout mouse, where, ablation of RXRγ was found to lead to depressive-like behavior in the mice16,17.
Moreover, chronic treatment with isoretinoin, the 13-cis-isomer and prodrug of retinoic acid that is used in acne therapy, is well known to cause severe depressive symptoms including suicide18. Given the tight regulation of RA homeostasis by local synthesis and differential local degradation via fine-tuned feedback mechanisms, it is not surprising that opposite effects can be observed for chronic, high-dose treatment with retinoids (inducing depressive-like behavior) and for acute, short-term treatment, which was reported to have antidepressive effects similar to those observed for fluoxetine17.
During embryonic development, a negative feedback was shown for RA on the expression of ROLDHs and thereby its own synthesis19. In almost all other developing or adult tissues, local degradation of RA via CYP450-enzymes is positively feedback-regulated by RA with RAR involvement20,21. Several isozymes of CYP450, including retinoid specific CYP26 isozymes but also less well characterized ones, have been identified to be involved in RA-catabolism22,23. While in adults CYP26A1 is mainly expressed in the liver, CYP26B1 has been shown to have the highest levels in the CNS24,25. Highlighting the clinical impact of the CYP450-system in the context of MDD, an association has been found between functionally relevant polymorphisms of RA-catabolizing CYP450-isozymes, such as CYP2C19, on the one hand, and response to antidepressant treatment26 as well as prevalence of depressive symptoms and risk for depression and suicide itself on the other hand27,28. Consequently, transgenic mice expressing the mutant human CYP2C19 gene were found to exhibit hippocampal atrophy and depressive-like symptoms29. Against the background of RA´s involvement in MDD pathogenesis and its catabolism via CYP450-isozymes, RA signaling might represent a missing link between pathologically increased catabolism (e.g. via aberrant CYP2C19 isoforms) and altered state of mood and behavior.
Common targets of fluoxetine and RA in MDD
Fluoxetine and RA exhibit several effects in common on MDD related processes, supporting the hypothesis of enhanced RA signaling as an underlying mechanism for fluoxetine’s serotonin-independent pleiotropic effects. Synaptic plasticity including structural and functional processes in neurons due to environmental influences such as injury, stress and inflammation, is well known to be affected during MDD pathogenesis31,32. Interestingly, RA signaling can directly affect homeostatic synaptic plasticity via RARα and the Calcium-dependent protein phosphatase calcineurin (CaN)33. Altered CaN function has also been linked to psychiatric disorders34, and CaN activity has been shown to be upregulated in murine hippocampus after chronic fluoxetine treatment. Moreover, upregulation of CaN leads to a sensibilisation of mice to the behavioral effects of fluoxetine35, overexpression of human CaN upstream regulator of calcineurin 1 (RCAN1) results in increased anxiety and RCAN1 knock-out mice show a lack of early fluoxetine response36. Taken together, these results point towards an involvement of CaN (and potentional downstream RA signaling) in fluoxetine’s mode of action. This pathway might explain fluoxetine’s capability to potently modulate neuronal plasticity37,38 as well as its neuroprotectant properties39,40. Conversely, chronic RA treatment, which likely results in dysbalance of the homeostatically controlled endogenous RA signaling, has been shown to suppress hippocampal neurogenesis in adult rats, being associated with increased hippocampal RARα expression and occurrence of depressive-like symptoms41.
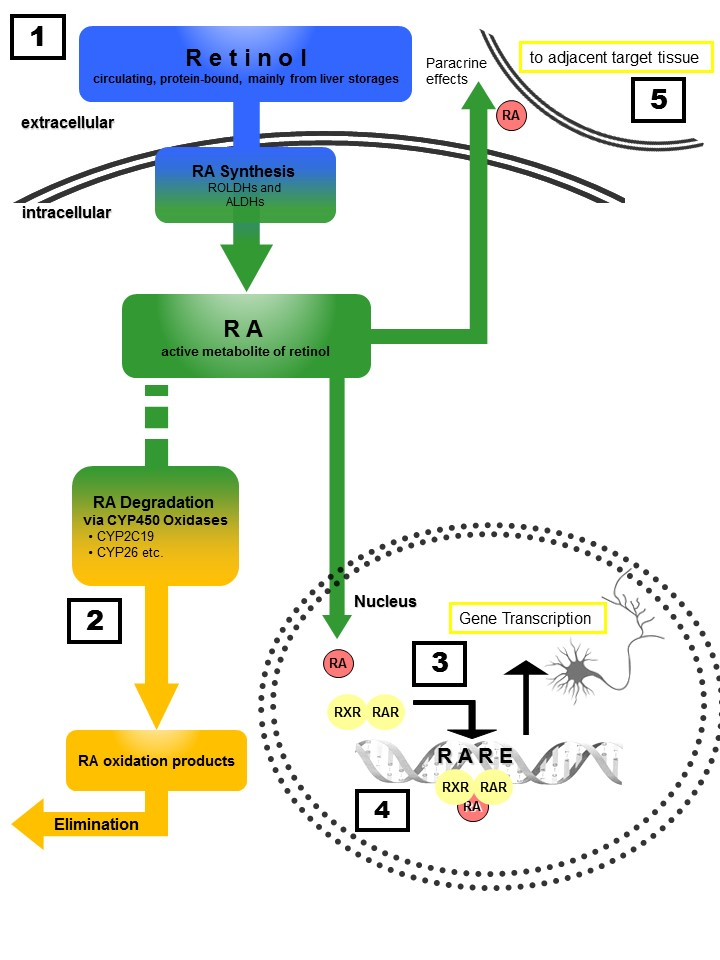
Figure 1: Retinoid homeostasis and involvement in MDD-related processes. Retinol, which is stored mainly in the liver, is released to the bloodstream bound to retinoid-binding proteins (RBPs). After uptake into target cells, retinol is oxidized via retinol dehydrogenases (ROLDH) and retinaldehyde dehydrogenases (RALDH) to retinoic acid (RA). RA then enters the nucleus of the same cell (autocrine actions) or diffuses to the nuclei of adjacent cells (paracrine actions) and binds to RXR and RAR receptors, forming homo- and heterodimers that induce transcription after binding to retinoic acid response elements (RARE). Chronic treatment with RA or its 13-cis isomer isoretinoin can lead to severe depressive symptoms, whereas acute RA treatment exhibits antidepressant effects similar to those observed for fluoxetine (1). Polymorphisms of RA-catabolizing CYP450-isozymes (2) are associated with response to antidepressants as well as the risk for developing depression. RA-signaling controls dopamine d2 receptor (D2R) expression (4) with D2R-upregulation resulting in antidepressant effects. Moreover, RA directly affects synaptic plasticity via RARα -mediated signaling (3), a process essentially involved in MDD pathogenesis.Furthermore, RA transcriptionally regulates CRH expression, with chronic RA exposure resulting in enhanced activity of the hypothalamic–pituitary–adrenal (HPA) axis. Finally, paracrine signaling (5) to adjacent cells results in anti-neuroinflammatory and neuroprotective actions.
Finally, MDD pathogenesis is strongly linked to chronic neuroinflammation, a process that is well inhibited by both, RA42,43 and fluoxetine5,44, providing yet another important overlap of RA- and fluoxetine-mediated effects. Another overlap between RA’s and fluoxetine’s anti-inflammatory properties has been noted at the level of cytokine release from astrocytes43 and overall inhibition of microglial activationas assessed by the release of the proinflammatory cytokine TNF-α42 or IL-645, demonstrating anti-inflammatory effects for both, fluoxetine and RA. With respect to the canonical chronic stress model for MDD pathogenesis, RA also seems to be involved: While hyperactivity of the CRH-driven hypothalamic–pituitary–adrenal (HPA) system is a common feature in some depressive patients, RA-signaling has been shown to control CRH gene expression directly via RARα-dependent signal transduction. In a rat model it has been shown that RA-exposure can further result in altered glucocorticoid receptor (GR) expression46 and overall enhanced HPA axis activity47. Fluoxetine is also known to alter GR expression and to change HPA activity via feedback–mechanisms48, providing yet another overlap between the neurobiological effects of RA and of fluoxetine. Another direct, likely serotonin-independent effect of fluoxetine was shown for the acid sphingomyelinase-ceramide (ASM) system. ASM is a glycoprotein that catalyzes sphingomyelin degradation into ceramides that, in higher concentrations, are associated with depressive symptoms. While fluoxetine can reduce ASM activity and, consequently, lower hippocampal ceramide concentrations7, RA has also been demonstrated to affect ASM-activity and lower ceramide levels, although lower ceramide levels have been attributed to increased ceramide degradation by ceramide kinase in RA treatment49.50. This provides another common target for RA and fluoxetine.. Against the background of the complex regulation underlying local RA signaling in the adult brain, it becomes clear that there is a pronounced difference between chronic RA-treatment18, which can increase depressive symptoms, and acute RA exposure, which reduces depressive symptoms17. This may point towards adaptational processes during long-term treatment, involving the tightly regulated feedback machinery that is meant to keep local RA signaling in a physiological balance. Such adaptational processes may include downregulation of RA synthesizing-and upregulation of RA degrading enzymes in a temporo-spatial manner, which in the long term may lead to effects opposite to thje effects seen for acute stimulation of RA signaling. Another highly relevant MDD-related target for RA signaling is the dopamine d2 receptor (D2R). RXRγ controls the expression of the canonical D2R, which is a relevant antidepressant target by numerous established antidepressant pharmacotherapies and known to be upregulated by chronic fluoxetine treatment16. Taken together, both fluoxetine and RA target common neuroprotective and pro-differentiative pathways, including key homeostatic and depression-related processes, suggesting a mechanism of action for fluoxetine that may potentially involve altered endogenous retinoid signaling (Figure 2).
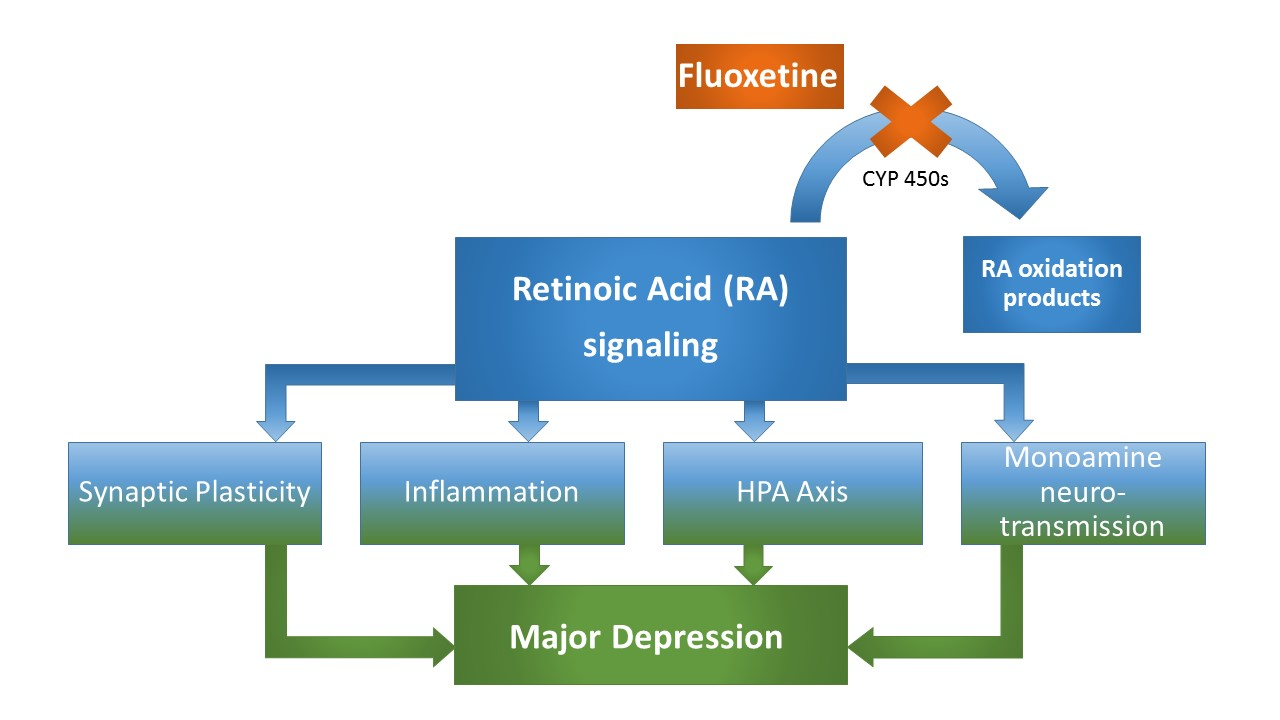
Figure 2: MDD-related neurobiological processes targeted by both, RA and fluoxetine. These processes include synaptic plasticity, that is directly affected by RA signaling at the level of homeostastic synaptic plasticity involving differential, calcium-triggered local RA synthesis, which essentially involves the calcium-dependent protein phosphatase calcineurin (CaN), a protein for which expression levels have been demonstrated to be affected by fluoxetine treatment and associated with fluoxetine response. Another common target of RA and fluoxetine is their anti-inflammatory property involving altered cytokine expression patterns. Moreover, both RA and fluoxetine alter HPA axis activity, while a precise mechanism involving RA-mediated CRH-transcription has been suggested. Finally, the canonical monoamine system is affected by both substances, with fluoxetine treatment resulting in an upregulation of the MDD-associated D2R, which is at the same time a known target of RA, bearing a retinoic acid response element in its promotor region. Taken together, numerous depression-related processes that are altered through fluoxetine treatment are equally classical targets of RA-signaling, strongly supporting a RA-dependent mechanism in fluoxetine’s pleiotropic mode of action (Figure 2).
Fluoxetine blocks RA degradation
Against the background of the common MDD-related pathways targeted by both, fluoxetine and RA, we recently hypothesized a direct interaction between fluoxetine and RA at a local pharmacokinetic level. Using traditional pharmacological approaches , we were able to show that degradation of RA can be potently inhibited by fluoxetine in a dose-dependent manner in brain tissue ex vivo, resulting in a a dose-dependent increase of RA (tissue-) levels. At a functional level, we were able to demonstrate that fluoxetine was able to reduce glutamate excitotoxicity on primary neurons to a similar extent observed for RA treatment. This effect was blocked by inhibition of RXRs, but augmented by a pan-RAR blocking agent, suggesting that RA signaling via RXR receptors may predominantly mediate the neuroprotective effects of fluoxetine8.
An interaction of RA and fluoxetine has also been suggested at the clinical level, where administration of fluoxetine has led to significant improvement of depressive symptoms after isoretinoin treatment in case reports18.
Taken together, there are several independent lines of evidence pointing towards RA-involvement in fluoxetine’s pleiotropic actions strongly supporting the experimentally demonstratedsupport a direct inhibition of RA degradation by fluoxetine to mediate its pleiotropic neuroprotective and anti-inflammatory mode of action.
References
- Whiteford HA, Degenhardt L, Rehm J, Baxter AJ, Ferrari AJ, Erskine HE, et al. Global burden of disease attributable to mental and substance use disorders: findings from the Global Burden of Disease Study 2010. Lancet (London, England). 2013;382(9904):1575-1586.
- Coppen A. The biochemistry of affective disorders. Br J Psychiatry. 1967;113(504):1237-1264.
- David DJ, Samuels BA, Rainer Q, Wang JW, Marsteller D, Mendez I, et al. Neurogenesis-dependent and -independent effects of fluoxetine in an animal model of anxiety/depression. Neuron. 2009;62(4):479-493.
- Malberg JE, Duman RS. Cell proliferation in adult hippocampus is decreased by inescapable stress: reversal by fluoxetine treatment. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology. 2003;28(9):1562-1571.
- Blatteau JE, de Maistre S, Lambrechts K, Abraini J, Risso JJ, Vallee N. Fluoxetine stimulates anti-inflammatory IL-10 cytokine production and attenuates sensory deficits in a rat model of decompression sickness. Journal of applied physiology (Bethesda, Md : 1985). 2015;119(12):1393-1399.
- Kornhuber J, Tripal P, Reichel M, Muhle C, Rhein C, Muehlbacher M, et al. Functional Inhibitors of Acid Sphingomyelinase (FIASMAs): a novel pharmacological group of drugs with broad clinical applications. Cellular physiology and biochemistry : international journal of experimental cellular physiology, biochemistry, and pharmacology. 2010;26(1):9-20.
- Gulbins E, Palmada M, Reichel M, Luth A, Bohmer C, Amato D, et al. Acid sphingomyelinase-ceramide system mediates effects of antidepressant drugs. Nature medicine. 2013;19(7):934-938.
- Hellmann-Regen J, Uhlemann R, Regen F, Heuser I, Otte C, Endres M, et al. Direct inhibition of retinoic acid catabolism by fluoxetine. Journal of neural transmission (Vienna, Austria : 1996). 2015;122(9):1329-1338.
- Maden M. Retinoic acid in the development, regeneration and maintenance of the nervous system. Nature reviews Neuroscience. 2007;8(10):755-765.
- Blomhoff R, Rasmussen M, Nilsson A, Norum KR, Berg T, Blaner WS, et al. Hepatic retinol metabolism. Distribution of retinoids, enzymes, and binding proteins in isolated rat liver cells. The Journal of biological chemistry. 1985;260(25):13560-13565.
- Kanai M, Raz A, Goodman DS. Retinol-binding protein: the transport protein for vitamin A in human plasma. The Journal of clinical investigation. 1968;47(9):2025-2044.
- Kawaguchi R, Yu J, Honda J, Hu J, Whitelegge J, Ping P, et al. A membrane receptor for retinol binding protein mediates cellular uptake of vitamin A. Science (New York, NY). 2007;315(5813):820-825
- Grunblatt E, Riederer P. Aldehyde dehydrogenase (ALDH) in Alzheimer's and Parkinson's disease. Journal of neural transmission (Vienna, Austria : 1996). 2016;123(2):83-90.
- Budhu AS, Noy N. Direct channeling of retinoic acid between cellular retinoic acid-binding protein II and retinoic acid receptor sensitizes mammary carcinoma cells to retinoic acid-induced growth arrest. Molecular and cellular biology. 2002;22(8):2632-2641.
- Leid M, Kastner P, Chambon P. Multiplicity generates diversity in the retinoic acid signalling pathways. Trends in biochemical sciences. 1992;17(10):427-433.
- Krzyzosiak A, Szyszka-Niagolov M, Wietrzych M, Gobaille S, Muramatsu S, Krezel W. Retinoid x receptor gamma control of affective behaviors involves dopaminergic signaling in mice. Neuron. 2010;66(6):908-920..
- Wietrzych-Schindler M, Szyszka-Niagolov M, Ohta K, Endo Y, Perez E, de Lera AR, et al. Retinoid x receptor gamma is implicated in docosahexaenoic acid modulation of despair behaviors and working memory in mice. Biological psychiatry. 2011;69(8):788-794.
- Bremner JD, Shearer KD, McCaffery PJ. Retinoic acid and affective disorders: the evidence for an association. The Journal of clinical psychiatry. 2012;73(1):37-50.
- Sandell LL, Lynn ML, Inman KE, McDowell W, Trainor PA. RDH10 oxidation of Vitamin A is a critical control step in synthesis of retinoic acid during mouse embryogenesis. PloS one. 2012;7(2):e30698.
- Pozzi S, Rossetti S, Bistulfi G, Sacchi N. RAR-mediated epigenetic control of the cytochrome P450 Cyp26a1 in embryocarcinoma cells. Oncogene. 2006;25(9):1400-1407.
- Rydeen A, Voisin N, D'Aniello E, Ravisankar P, Devignes C-S, Waxman JS. Excessive feedback of Cyp26a1 promotes cell non-autonomous loss of retinoic acid signaling. Developmental biology. 2015;405(1):47-55.
- McSorley LC, Daly AK. Identification of human cytochrome P450 isoforms that contribute to all-trans-retinoic acid 4-hydroxylation. Biochemical pharmacology. 2000;60(4):517-526.
- Marill J, Cresteil T, Lanotte M, Chabot GG. Identification of human cytochrome P450s involved in the formation of all-trans-retinoic acid principal metabolites. Molecular pharmacology. 2000;58(6):1341-1348.
- JThatcher JE, Isoherranen N. The role of CYP26 enzymes in retinoic acid clearance. Expert opinion on drug metabolism & toxicology. 2009;5(8):875-886.
- Topletz AR, Thatcher JE, Zelter A, Lutz JD, Tay S, Nelson WL, et al. Comparison of the function and expression of CYP26A1 and CYP26B1, the two retinoic acid hydroxylases. Biochemical pharmacology. 2012;83(1):149-163.
- Ruano G, Villagra D, Szarek B, Windemuth A, Kocherla M, Gorowski K, et al. Physiogenomic analysis of CYP450 drug metabolism correlates dyslipidemia with pharmacogenetic functional status in psychiatric patients. Biomarkers in medicine. 2011;5(4):439-449.
- Ingelman-Sundberg M, Persson A, Jukic MM. Polymorphic expression of CYP2C19 and CYP2D6 in the developing and adult human brain causing variability in cognition, risk for depression and suicide: the search for the endogenous substrates. Pharmacogenomics. 2014;15(15):1841-1844.
- Sim SC, Nordin L, Andersson TM, Virding S, Olsson M, Pedersen NL, et al. Association between CYP2C19 polymorphism and depressive symptoms. American journal of medical genetics Part B, Neuropsychiatric genetics : the official publication of the International Society of Psychiatric Genetics. 2010;153b(6):1160-1166.
- Persson A, Sim SC, Virding S, Onishchenko N, Schulte G, Ingelman-Sundberg M. Decreased hippocampal volume and increased anxiety in a transgenic mouse model expressing the human CYP2C19 gene. Molecular psychiatry. 2014;19(6):733-741.
- Jeppesen U, Gram LF, Vistisen K, Loft S, Poulsen HE, Brosen K. Dose-dependent inhibition of CYP1A2, CYP2C19 and CYP2D6 by citalopram, fluoxetine, fluvoxamine and paroxetine. European journal of clinical pharmacology. 1996;51(1):73-78.
- Fossati P, Radtchenko A, Boyer P. Neuroplasticity: from MRI to depressive symptoms. European neuropsychopharmacology : the journal of the European College of Neuropsychopharmacology. 2004;14 Suppl 5:S503-510.
- . Duman RS. Pathophysiology of depression: the concept of synaptic plasticity. European psychiatry : the journal of the Association of European Psychiatrists. 2002;17 Suppl 3:306-310.
- Maghsoodi B, Poon MM, Nam CI, Aoto J, Ting P, Chen L. Retinoic acid regulates RARalpha-mediated control of translation in dendritic RNA granules during homeostatic synaptic plasticity. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(41):16015-16020.
- Gerber DJ, Hall D, Miyakawa T, Demars S, Gogos JA, Karayiorgou M, et al. Evidence for association of schizophrenia with genetic variation in the 8p21.3 gene, PPP3CC, encoding the calcineurin gamma subunit. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(15):8993-8998.
- Crozatier C, Farley S, Mansuy IM, Dumas S, Giros B, Tzavara ET. Calcineurin (protein phosphatase 2B) is involved in the mechanisms of action of antidepressants. Neuroscience. 2007;144(4):1470-1476.
- Hoeffer CA, Wong H, Cain P, Levenga J, Cowansage KK, Choi Y, et al. Regulator of calcineurin 1 modulates expression of innate anxiety and anxiogenic responses to selective serotonin reuptake inhibitor treatment. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2013;33(43):16930-16944.
- Maya Vetencourt JF, Sale A, Viegi A, Baroncelli L, De Pasquale R, O'Leary OF, et al. The antidepressant fluoxetine restores plasticity in the adult visual cortex. Science (New York, NY). 2008;320(5874):385-388.
- Zhou Y, Notterpek L. Promoting peripheral myelin repair. Exp Neurol. 2016.
- Chollet F, Tardy J, Albucher JF, Thalamas C, Berard E, Lamy C, et al. Fluoxetine for motor recovery after acute ischaemic stroke (FLAME): a randomised placebo-controlled trial. The Lancet Neurology. 2011;10(2):123-130
- Lim CM, Kim SW, Park JY, Kim C, Yoon SH, Lee JK. Fluoxetine affords robust neuroprotection in the postischemic brain via its anti-inflammatory effect. Journal of neuroscience research. 2009;87(4):1037-1045.
- Hu P, Wang Y, Liu J, Meng F-T, Qi X-R, Chen L, et al. Chronic retinoic acid treatment suppresses adult hippocampal neurogenesis, in close correlation with depressive-like behavior. Hippocampus. 2016:n/a-n/a.
- Hellmann-Regen J, Kronenberg G, Uhlemann R, Freyer D, Endres M, Gertz K. Accelerated degradation of retinoic acid by activated microglia. Journal of neuroimmunology. 2013;256(1-2):1-6.
- van Neerven S, Nemes A, Imholz P, Regen T, Denecke B, Johann S, et al. Inflammatory cytokine release of astrocytes in vitro is reduced by all-trans retinoic acid. Journal of neuroimmunology. 2010;229(1-2):169-179.
- Kao CY, He Z, Zannas AS, Hahn O, Kuhne C, Reichel JM, et al. Fluoxetine treatment prevents the inflammatory response in a mouse model of posttraumatic stress disorder. Journal of psychiatric research. 2016;76:74-83.
- Blatteau JE, Barre S, Pascual A, Castagna O, Abraini JH, Risso JJ, et al. Protective effects of fluoxetine on decompression sickness in mice. PloS one. 2012;7(11):e49069.
- Hu P, Liu J, Zhao J, Qi XR, Qi CC, Lucassen PJ, et al. All-trans retinoic acid-induced hypothalamus-pituitary-adrenal hyperactivity involves glucocorticoid receptor dysregulation. Translational psychiatry. 2013;3:e336.
- Cai L, Yan XB, Chen XN, Meng QY, Zhou JN. Chronic all-trans retinoic acid administration induced hyperactivity of HPA axis and behavioral changes in young rats. European neuropsychopharmacology : the journal of the European College of Neuropsychopharmacology. 2010;20(12):839-847.
- Heydendael W, Jacobson L. Widespread hypothalamic-pituitary-adrenocortical axis-relevant and mood-relevant effects of chronic fluoxetine treatment on glucocorticoid receptor gene expression in mice. The European journal of neuroscience. 2010;31(5):892-902.
- Murakami M, Ito H, Hagiwara K, Yoshida K, Sobue S, Ichihara M, et al. ATRA inhibits ceramide kinase transcription in a human neuroblastoma cell line, SH-SY5Y cells: the role of COUP-TFI. Journal of neurochemistry. 2010;112(2):511-520.
- Murate T, Suzuki M, Hattori M, Takagi A, Kojima T, Tanizawa T, et al. Up-regulation of acid sphingomyelinase during retinoic acid-induced myeloid differentiation of NB4, a human acute promyelocytic leukemia cell line. The Journal of biological chemistry. 2002;277(12):9936-9943.