
Riikka-Liisa Uronen and Henri J. Huttunen*
Neuroscience Center, University of Helsinki, FI-00014 Helsinki, Finland
In Alzheimer’s disease (AD), loss of neurons and synapses parallels the formation of neurofibrillary tangles, protein aggregates mainly composed of hyperphosphorylated and aggregated Tau protein. Tau is mostly a cytosolic protein but can also be secreted by neurons. Cell-to-cell transfer of misfolded Tau protein plays a key role in the spread of neurofibrillary pathology between brain regions in AD and other tauopathies. Advances in genome-wide technologies have identified a large number of genetic risk factors for late-onset AD (LOAD). Currently, it remains unknown if genetic factors influence disease risk or progression rate by altering cell-to-cell propagation of Tau. Several LOAD risk genes are functionally associated with endocytic trafficking providing a potential link to Tau secretion and uptake. Recently, a LOAD risk gene FRMD4A was shown to regulate Tau secretion via a pathway linked to presynaptic vesicle machinery and polarity signaling. Tau release is linked to neuronal activity, and genetic factors that affect presynaptic vesicle release in the aging brain may also influence disease progression in AD and other tauopathies. In this mini review, we summarize the recent literature with a focus on the role of FRMD4A-cytohesin-Arf6 pathway and presynaptic vesicle machinery in the secretion of Tau.
Tau pathology in AD progresses through anatomically connected brain regions, beginning in the transentorhinal region, then involving the hippocampus and finally the neocortex1. The number and distribution of neurofibrillary tangles (NFTs) is strongly correlated with progressive cognitive loss in AD and most other tauopathies.
Tau, a cytosolic microtubule-associated protein, can be released from cells, including neurons, under physiological conditions2. Misfolded and aggregated Tau has been reported to transfer between neighboring cells in vitro3-5 and in vivo6-8. Multiple other proteins associated with neurodegenerative diseases, including amyloid-β peptide (Aβ), α-synuclein, mutant SOD1, mutant huntingtin and TDP-43, have also been shown to propagate protein misfolding pathology between cells in a similar prion-like manner, although the respective neuropathological cascades may vary in the human brain (reviewed in9,10). How the accumulation and spreading of Aβ and Tau aggregates are mechanistically connected in human AD brain remains largely a mystery11. Clearly, however, Tau pathology can develop and spread in the absence of Aβ plaques, as evidenced by numerous tauopathies, such as familial cases of frontotemporal dementia that are primarily driven by aggregation-promoting mutations in the MAPT gene (that encodes the Tau protein)12. Also, there is an on-going debate whether primary age-related tauopathy (PART), an Aβ-independent temporal lobe NFT pathology frequently observed in the brains of aged individuals, is a separate disease entity from AD13,14.
While the cell-to-cell transmission paradigm now appears to explain how the neurodegenerative pathology spreads between brain regions, the cellular mechanisms of secretion and uptake of misfolded intracellular proteins remain incompletely understood. Conformational templating, a characteristic of amyloids in general, drives the seeding and accumulation of pathological protein aggregates9,10. Interestingly, specific conformations and strains with distinct propagation properties have been described for Tau5. It is currently unclear whether the release of Tau species from neurons is related to a normal physiological release pathway, unconventional secretory mechanisms or mechanisms related to neuronal injury. Cellular uptake of Tau fibrils has been shown to involve binding to heparan sulphate proteoglycans on the cell surface followed by macropinocytosis15, an actin-dependent endocytic process that allows the entry of fluid-phase macromolecular structures into the cell. However, membrane turnover in neurons is tightly controlled, and basal macropinocytic activity in mature neurons appears to be rather low16. Additional stimulation, such as axonal injury, may be required to promote neuronal uptake of protein aggregates via macropinocytosis. Importantly, microglial cells facilitate Tau propagation in vivo by packing phagocytosed Tau to exosomes that are more effectively uptaken by neurons than vesicle-free Tau species17.
Early-onset AD is an almost entirely genetically determined disease, characterized by highly penetrant disease-causing mutations in three genes (APP, PSEN1 and PSEN2) that are all functionally linked to generation of amyloid-β peptide18. In contrast, LOAD is a complex disorder with heterogeneous etiology, and variable age of onset and progression rate. The APOE gene (encoding apolipoprotein E) is a major genetic risk factor for LOAD19. Genome-wide association studies (GWAS) have identified a large number of common risk variants within >20 genetic loci associated with LOAD20-25, including CLU, ABCA7, CD2AP, BIN1, CR1, CD33 and the MS4A gene cluster 20-25. Next-generation sequencing efforts have identified additional rare variants with strong effects on disease risk, including TREM2 and ABCA726-29. In addition, genome-wide association studies have linked FRMD4A gene to LOAD21 and late-life cognitive decline30. Although the exact functional roles of individual susceptibility genes remain poorly understood, the main LOAD-associated genetic loci appear to be functionally linked to three major biological pathways: immune system, lipid metabolism and cell membrane processes (e.g. endocytosis, synaptic function)31,32. Significantly increased levels of Tau in the cerebrospinal fluid (CSF) are associated with faster rate of cognitive decline and overall worse clinical outcome in AD33,34. In general, alleles associated with lower CSF Tau levels would thus be considered protective for disease risk, associated with less tau pathology and with slower cognitive decline, and vice versa. In support of this, genetic variants linked to increased CSF phospho-Tau (Thr181) levels were also associated with faster rate of disease progression while having no effect on disease risk or age of onset36. In particular, a single-nucleotide polymorphism (SNP; rs1868402) in the PPP3R1 gene linked to reduced parietal lobe expression of protein phosphatase B (calcineurin), a known Tau phosphatase, was associated with higher CSF phospho-Tau levels and faster rate of disease progression. Another genome-wide association study that used CSF Tau levels as an endophenotype of LOAD identified novel risk variants of the disease, including SNAR-I, GLIS3 and TOMM40, and confirmed the association of APOE and TREM2 with the variability of CSF Tau and phospho-Tau levels36. These studies clearly indicate that common genetic variants have an impact on the CSF levels of Tau and that this may be associated with the risk of LOAD or rate of disease progression. Interestingly, none of the SNPs linked to altered CSF levels of Tau were associated with Tau (MAPT) expression levels suggesting that they affect CSF Tau levels by a post-transcriptional mechanism, which could potentially include mechanisms regulating cellular release and uptake of Tau.
Despite the rapidly accumulating genetic data, there remains a knowledge gap regarding the functional association of the risk genes and gene variants to the pathobiology of complex diseases. Single nucleotide polymorphisms that act as expression quantitative trait loci (eQTL) influencing gene expression constitute an important class of functional variants of genes. Several LOAD-associated risk variants, such as CLU, MS4A4A and ABCA7, harbor eQTLs37. Recently, transcriptional analysis of Braak-staged temporal cortex samples from AD patients and healthy controls revealed that the expression of FRMD4A, MS4A6A, CLU and TREM2 was altered in relation to increasing AD-related neurofibrillary pathology38.
To gain more insight into the functional roles of LOAD risk genes, we combined individual silencing of selected LOAD risk genes (APOE, BIN1, CLU, ABCA7, CR1, PICALM, CD33, CD2AP, FRMD4A and TREM2; based on LOAD genetics meta-analyses39 and recent literature), together with AD pathobiology-based pathway analysis. We previously developed a panel of sensitive live-cell assays for monitoring changes of pathologically central protein-protein interactions in AD, such as key protein interactions related to Aβ generation and Tau hyperphosphorylation38,40,41,42. In combination with RNAi silencing of LOAD susceptibility genes, the AD-specific pathway sensors provide an easily accessible platform for studying functional roles of LOAD risk genes in live cells.
While the expression of FRMD4A was found to be decreased in relation to increasing neurofibrillary pathology in the temporal cortex of LOAD patients, in vitro pathway analysis showed that reduced FRMD4A expression associates with both increased amyloidogenic amyloid precursor protein (APP) processing and increased Tau phosphorylation activity38. Furthermore, our recent study showed that altered FRMD4A level also significantly alters Tau secretion41, for the first time functionally linking a LOAD risk gene to basic cellular mechanisms of cell-to-cell transfer of Tau. None of the other top-ten LOAD risk genes included in this study showed a functional connection to Tau secretion. APOE knockdown caused a subtle increase in cellular uptake of Tau, an effect possibly related to the direct interaction of ApoE and Tau proteins43 in the extracellular space.
FRMD4A (FERM Domain Containing 4A) protein is involved in polarization of epithelial cells44 and mutations in the FRMD4A gene lead to microcephaly and mental retardation in humans45. However, most of the physiological functions of FRMD4A are so far unclear. Our data clearly suggests that FRMD4A-cytohesin-Arf6 pathway regulates Tau secretion41. Activation of this pathway in HEK293T cells by overexpressing FRMD4A or Arf6 leads to increased Tau secretion, whereas inhibition by FRMD4A RNAi or cytohesin inhibitor SecinH3 decreased it. Surprisingly, this effect was opposite in cortical neuron cultures, where inhibition of this pathway lead to an increase in Tau secretion. The significant increase of neuronal Tau secretion by FRMD4A/cytohesin inhibition suggests that the reduced FRMD4A levels in AD brain may be causally linked to spreading of Tau pathology.
Cytohesinsa are a family of Arf Guanine Nucleotide Exchange Factors (GEFs) that have a central Sec7-GEF-domain, a protein-protein interaction mediating coiled coil (CC) domain and a membrane targeting pleckstrin homology (PH) domain. Cytohesins bind FRMD4A44 and activate the small GTPase Arf646 promoting its translocation to the plasma membrane47. Cytohesins function in various cellular processes including insulin receptor signaling48, integrin trafficking and cell migration46. Importantly, they also regulate neurotransmission47 and membrane trafficking49 at the presynaptic terminal, probably via their interaction with Munc13s50, that are proteins essential for synaptic vesicle priming51. As Tau secretion is related to neurotransmitter release2,52, it seems plausible that the changes we see in Tau secretion with modulation of FRMD4A-cytohesin pathway are connected to synaptic vesicle release. The extreme specialization of the presynaptic terminal may also explain why the effect of the FRMD4A-cytohesin pathway is different in neurons and in HEK293T cells51.
1Cytohesin family aliases:
Cytohesin 1 = mSec7-1, PSCD-1,
Cytohesin 2 = mSec7-2, ARNO, PSCD-2
Cytohesin 3 = mSec7-3, GRP-1, PSCD-3
Cytohesin 4 = PSCD-4
The polarity signaling complex Par3/Par6/aPKCζ (Partitioning defective 3, Partitioning defective 6 and atypical Protein Kinase C ζ, respectively), that associates with FRMD4A and regulates its activity in epithelial cells44 also affects Tau secretion41 (Figure 1). To our knowledge this is the first study implicating a link between cell polarity signaling and Tau secretion. As neurons are highly polarized cells, polarity signaling is not only important for their development and differentiation53 but also may play a role in plasticity54. In addition, polarity signaling proteins have a more general role in endocytic trafficking55, which may also lead to changes in Tau release.
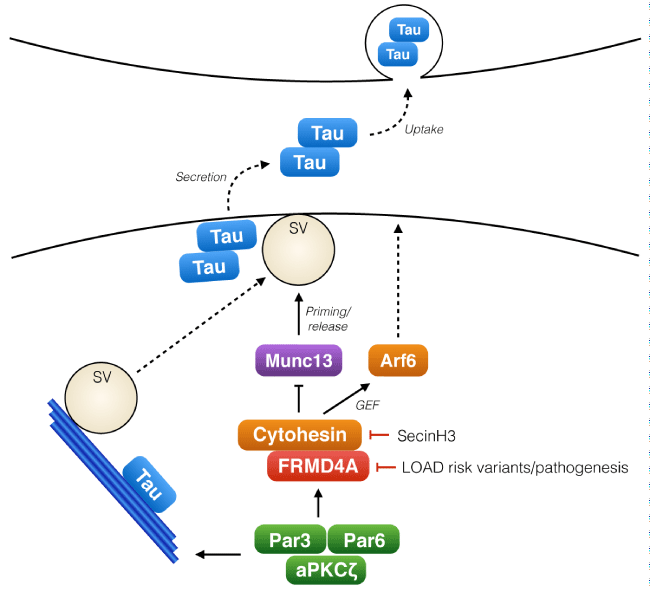
Figure 1: The role of FRMD4A-cytohesin-Arf6 pathway in presynaptic release of Tau.
Tau, a cytosolic microtubule regulating protein, is released from cells in association with neuronal activity. The polarity signaling complex Par3/Par6/aPKCζ regulates the activity of FRMD4A and cytohesins, which upon activation promote membrane localization of Arf6 and also interact with Munc13s, key regulators of synaptic vesicle (SV) priming. Activity of this signaling module regulates the release of Tau from the presynaptic terminal to the extracellular space. Tau release is enhanced by reduced expression of FRMD4A (RNA interference mimicking the decreased FRMD4A levels in LOAD patients) or with the cytohesin inhibitor SecinH3.
In addition to Tau phosphorylation38 and secretion41, FRMD4A also affects Aβ secretion38. Notably, the secretion of Aβ into the extracellular space is also tightly regulated by neuronal activity in vitro56 and in vivo557. Whether Aβ secretion is similarly regulated with cytohesin/Arf6 and Par3/Par6/aPKCζ remains to be studied. Interestingly, cytohesins were recently also associated with another neurodegenerative disease, amyotrophic lateral sclerosis (ALS)58, but whether this is related to our findings needs to be further investigated.
LOAD risk genes involved in endocytic trafficking, synaptic function and microglial activity may affect AD risk and progression through regulation of cellular secretion and uptake of Tau. The expression of LOAD risk gene FRMD4A is decreased in Alzheimer’s disease patient brains, and a decrease in FRMD4A protein level leads to an increase of neuronal Tau secretion. Based on these results, we suggest that a presynaptic signaling module consisting of FRMD4A, cytohesins and Arf6 acts as a regulator of cell-to-cell transmission of Tau.
LOAD risk genes involved in endocytic trafficking, synaptic function and microglial activity may affect AD risk and progression through regulation of cellular secretion and uptake of Tau. The expression of LOAD risk gene FRMD4A is decreased in Alzheimer’s disease patient brains, and a decrease in FRMD4A protein level leads to an increase of neuronal Tau secretion. Based on these results, we suggest that a presynaptic signaling module consisting of FRMD4A, cytohesins and Arf6 acts as a regulator of cell-to-cell transmission of Tau.