
Karenitecin (bnp1350) and flavopridol as radiosensitizers in malignant glioma
Deepika Rajesh, H. Ian Robins*, Steven P. Howard
Abstract
The poor prognosis of malignant glioma patients highlights the need to develop low toxicity, tumor specific agents with the ability to synergize with proven efficacious treatment modalities, e.g., ionizing irradiation. This paper investigates the potential of BNP1350 (karenitecin), a topoisomerase I-targeting anticancer agent, and flavopridol a cyclin-dependent kinase inhibitor as radiosensitizers at clinically relevant doses in glioblastoma cell lines. A clonogenic survival and apoptosis assays were performed to test the effect of karenitecin (0.1 nM to 10 nM), flavopridol, (50 nM to 500 nM), radiation (1 Gy to 5.5 Gy) and a combination of radiation and karenitecin or radiation and flavopridol on the glioma cell lines T986 and M059K. Cells were stained for cyclins B and D using antibodies followed by flow cytometry. Propidium Iodide staining was used to reveal the various phases of the cell cycle; cyclin staining in the G0/G1 and G2/M phase of the cell cycle was estimated as the Mean Fluorescence Intensity (MFI) after subtracting the MFI recorded by the isotype controls. Results demonstrated that in irradiated cells, pretreatment with karenitecin induced apoptosis, a transient arrest in the G2/M phase of the cell cycle and increased the expression of cyclin B1. Flavopridol treatment also induced apoptosis and a transient block in the G2/M phase of the cell cycle. The combined effects of karenitecin and flavopridol displayed synergistic effects. The unique radiosensitizing activity of orally administrable karenitecin and flavopridol is consistent with continued investigation of these compounds preclinically, as well as in the clinical setting.
Abbreviations:
Cyclin-dependent kinase (CDK); blood brain barrier (BBB); room temperature (RT); Fluorescein Isothiocynate (FITC)
Introduction
Surgery and radiotherapy currently remains the mainstay of treatment for high-grade gliomas. Advances in the last decade have included technological advances in both of these modalities, as well as the introduction of chemotherapy, e.g., temozolomide. Interestingly, the vast majority of recurrences (in radiation treated patients) will ultimately develop immediately within or in proximity to initial treatment fields. Thus, the development of an effective radiosensitizer is particularly relevant in the management of this poor prognosis patient group. As described below we elected to investigate two potential targets for inducing radiosensitization, i.e., topoisomerase1 and CDK. In selecting suitable inhibitors, parameters included potential clinical tolerability, and ability to cross the blood brain barrier (BBB).
Flavopridol
It has been shown that flavopridol [5,7-dihydroxy-8-(4-N-methyl-2-hydroxypyridyl)-6'-chloroflavone hydrochloride, L86–8275, HMR 1275] is a potent and specific inhibitor of cyclin dependent kinases (CDK)s 1, 2, 4 and 7, receptor tyrosine kinase (epidermal growth factor receptor) and signal transducing kinases CPK1 and Erk1, and has undergone clinical testing in phase I and phase II trials1-9. In this regard, the efficacy of many antitumor agents is dependent on cell proliferation10. Relative to this, cell cycle progression is controlled by the activity of CDKs, which in turn are regulated by cyclin accumulation, by positive and negative phosphorylation events, and by association with inhibitory proteins11,12. Two major classes of CDK inhibitors have been identified in mammalian cells and are referred to as the INK and KIP families13. The INK family member p1610INK-4a blocks progression through G1 by inhibiting cyclin D kinases and plays a crucial role in cellular senescence13-15. The KIP-type inhibitors p21WAF-1 and p27Kip-1 regulate the progression from G1 into the S phase by inhibiting cyclin E kinase activity. p21WAF-1 is instrumental in invoking a G1 arrest following DNA damage or perturbation of nucleotide metabolism13. Regulation of CDKs is altered in a number of tumor types, and therefore competitive CDK inhibitors targeting specifically to the ATP binding cleft are a particularly attractive target group of drugs that could be used in cancer treatment.
In summary, flavopiridol is a synthetic flavone, which has previously been evaluated as an antineoplastic agent6-8,16-21. It is a well-tolerated drug; the dose used in clinical trials ranges between 0.1-0.2 mg/ml. It induces growth arrest at either the G1 and/or G2phases of the cell cycle in numerous cell lines in vitro by acting as a competitive binding agent for the ATP-binding pocket of CDK22,23. Flavopridol has been reported to bind to duplex DNA1,24. Flavopridol also inhibits receptor tyrosine kinases (EGFR), tyrosine kinases (pp60 Src) and signal transducing kinases (PKC and Erk-1)23,25. Although the inhibiting activity of flavopridol is strongest for CDK, the cytotoxic activity of flavopiridol is not limited to cycling cells as resting cells are also killed.
Karenitecin
Mammalian DNA topoisomerase I is the target of a number of active anticancer drugs known as camptothecins (e.g., topotecan and irinotecan). These topoisomerase inhibitors exert their cytotoxic effect by producing enzyme-mediated DNA damage, rather than by directly inhibiting enzyme catalytic activity. Recently, a series of novel camptothecin analogues, 7-silylcamptothecins ("silatecans"), have shown promising regression of U87 glioma cells in a nude mouse model and displayed lipophilicity to favor (BBB) transit26. Karenitecin a drug in this class (which has entered clinical trials) is a highly lipophilic, poorly water-soluble semisynthetic derivative of camptothecin, which can be administered orally. It displays increased stability at physiologic pH and has demonstrated cytotoxicity against human head and neck carcinoma and colon cancer cell lines27,28. The anti-tumor activity of karenitecin has been comparable of that of Topotecan in a xenograft model29,30. Grossman et al (2008)31 have concluded a phase 1 study in recurrent glioma patients with a maximum tolerated dose of 1.5 mg/m2 (and 2.0 mg/m2 in patients receiving enzyme-inducing anti-seizure drugs). The drug was well tolerated on a schedule of intravenous administration over 60 minutes daily for 5 days every 3 weeks. Median survival time after entering the study was 6.0 (95% CI 3.9 -9.7) months for 30 evaluable patients (23 glioblastoma; 7 anaplastic glioma).
The text that follows summarizes a preclinical investigation regarding the potential application of karenitecin and/or flavopridol as an adjunct to radiation treatment in malignant glioma cell lines.
Materials and methods
Materials
Karenitecin (BNP1350) was provided by Dr. Frederick H. Hausheer, Bionumerik Pharmaceuticals Inc., San Antonio, TX. Flavopridol was provided by Dr. Mark Ritter; University of Wisconsin Madison, Propidium Iodide (PI) and RNase H were purchased from Sigma Aldrich (St. Louis MO), antibodies to Cyclin B and D were purchased from Santa Cruz Biotechnology (Santa Cruz, CA), and Annexin staining kit was purchased from Clonetech (Palo Alto, CA).
Cell lines
The T98G32,33 and MO59K34 were obtained from ATCC and maintained in a humidified incubator with 5% CO2 at 37°C. T98G and MO59K cells were grown in DMEM F12 medium containing 10% fetal bovine serum, 1% penicillin, streptomycin and 1mM non-essential amino acids.
Cell treatment and clonogenic survival assays
The clonogenic survival assay was performed to test the effect of different doses of karenitecin (0.1nM to 10nM), flavopridol, (50nM to 500nM), radiation (1 Gy up to 8.5 Gy) and a combination of radiation and karenitecin or radiation and flavopridol on glioma cell lines. Stock solution of karenitecin was made in DMSO, and stock solution of flavopridol was made in PBS for all the experiments. Sub-confluent plates of glioma cell lines were treated with varying doses of karenitecin/flavopridol for a period of 72 hours. All cells were irradiated at a dose rate of approximately 7.5 Gy/min using a 137Cs irradiator. The dose of irradiation varied from 1 Gy up to 8.5 Gy. Cells treated with different doses of radiation or DMSO [<0.1%] served as control for karenitecin treated cells, while cells treated with different doses of radiation or medium alone served as control for karenitecin treated cells. Following irradiation, both drug treated and untreated cells were harvested; washed with PBS; plated at the desired cell number and incubated for two weeks. The colonies were stained with crystal violet and percentage survival was quantified. Survival was determined as the ratio of plating efficiencies for each irradiated group to that of the unirradiated control. For the flavopridol karenitecin combination experiments, the cells were pretreated with the indicated doses of either flavopridol or karenitecin; or a combination of both the drugs together for 72 hours followed by irradiation at 5.5 Gy. The cells were harvested, plated and the percentage survival was determined as described before
Apoptosis assays
The percent of cells undergoing apoptosis and the different phases of the cell cycle were quantified by the method described by Darzynkiewicz et al35,36. Briefly, both the cell lines were treated with karenitecin [0.1 nM – 10 nM or flavopridol], [50 nM to 500 nM] for a period of 24, 48 and 72 hours. For the karenitecin flavopridol combination experiments the cells were treated with 0.1 nM or 1 nM karenitecin with 100 nM flavopridol for 72 hours.
At appropriate times after treatment, the cells were harvested by trypsin-EDTA treatment and washed twice with ice-cold PBS. The cell pellet was resuspended in PBS and fixed in ice-cold 95% ethanol. The fixed cells were stained using a PI staining solution containing 20 ug/ml PI, 200 ug/ml RNase H and 0.1% NP40 solution. The cells were filtered through a 40µM pore nylon mesh (Tetkop, Inc.) and analyzed using a Beckton-Dickinson FACStar plus flow cytometry with excitation 488nM. The percentage of cells in sub-G0/G1 peak and the distribution of cells in G0/G1, S and G2/M phases were determined using the Modfit LT version 2.0 (Verity Software, Topham, Maine) and Cell Quest software (Becton Dickinson, San Jose, CA).
A clonogenic survival assay was performed to test the effect of different doses of karenitecin (0.1 nM to 10 nM), flavopridol, (50 nM to 500 nM), radiation (1 Gy up to 8.5 Gy) and a combination of radiation and karenitecin or radiation and flavopridol on glioma cell lines37. Cell lines were treated with varying doses of karenitecin, or flavopridol for the indicated times, harvested by using cell dissociation solution, washed and subsequently stained with antibody to annexin V conjugated to FITC and PI [10 µg/ml] using the Apoalert Kit (Clonetech, Palo Alto, CA) according to the manufacturer’s instructions. Viable [annexin V¯/PI¯] pre-apoptotic [annexin V+/PI¯], apoptotic [annexin V+/PI+], and the residual damaged [annexin V¯/PI+] cells are quantified using this protocol
Staining for Cyclins
Staining for cyclins was performed according to the method described by Darzynkiewicz et al38. Briefly, subconfluent cultures of T98G and MO59K cells were treated with the indicated concentrations karenitecin in the presence and absence of radiation for 72 hours. The cells were harvested and fixed in ice-cold ethanol overnight at 4° C. The cells were washed, centrifuged, and suspended in permeabilization solution (0.25%v/v Triton X-100 in PBS pH 7.4) and kept on ice for 5 minutes. The cells were washed again, resuspended in 100μl wash buffer (1% BSA in PBS); and incubated with antibodies to cyclins B, D or isotype control (Santa Cruz biotechnology, CA) for 60 minutes. The cells were washed, and stained with goat anti-mouse FITC for 30 minutes at RT. The stained cell preparation was stained with 5 µg/ml PI and 200 µg/ml DNase-free RNase A. The cells were analyzed on a FACScan flow cytometer (Becton Dickinson, San Jose, CA) and the results were acquired using CELL quest software and the Modfit LT version 2.0 (Verity Software, Topham, Maine). PI staining was used to reveal the various phases of the cell cycle and the cyclin staining in the G0/G1 and G2/M phase of the cell cycle was estimated as the MFI after subtracting the Mean Fluorescence Intensity recorded by the isotype control for each sample.
Results
Karenitecin induced radiosensitization in glioblastoma cell lines
The clonogenic survival assay was performed to determine the effect of different doses of karenitecin (0.1 nM – 10 nM), radiation (1-5.5 Gy) and a combination of both in T98G and MO59K cell lines. The results (Figures 1 and 2) revealed that pretreatment with karenitecin resulted in a dose dependent radiosensitization in the clinically relevant dose range of radiation. Higher concentrations of karenitecin between 10 – 100 nM proved toxic to the cells, and hence, radiosensitization experiments were performed in the dose range between 0.1 - 10 nM of karenitecin.
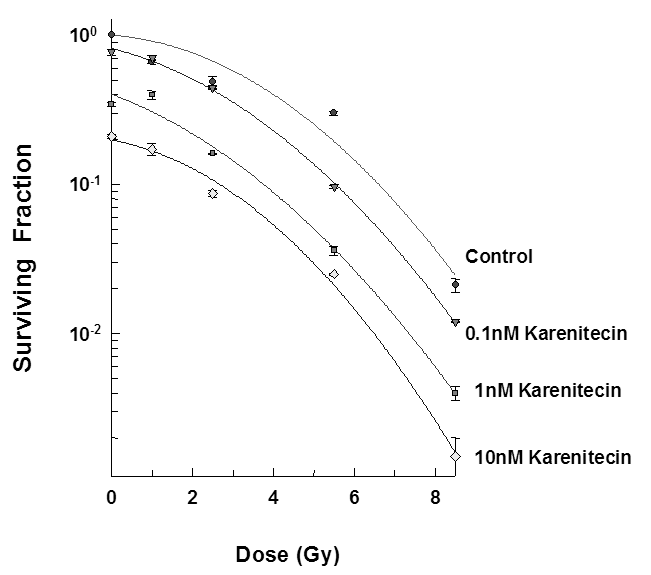
Figure 1: Subconfluent cultures of T98G cells (Figure 1) or MO59K (Figure 2) were treated with 0.1 nM, 1 nM and 10 nM concentrations of karenitecin for 72 hours and subjected to different doses of irradiation (1 Gy – 8.5 Gy). The control set of cells was treated with the indicated doses of radiation, or karenitecin, or with the vehicle control (DMSO) alone. The cells were harvested and an optimum number of cells were plated and allowed to grow for 14 days. The resulting colonies were stained and counted. Each graphed point represents mean values ± SE values of triplicate dishes (variation less than 5%).
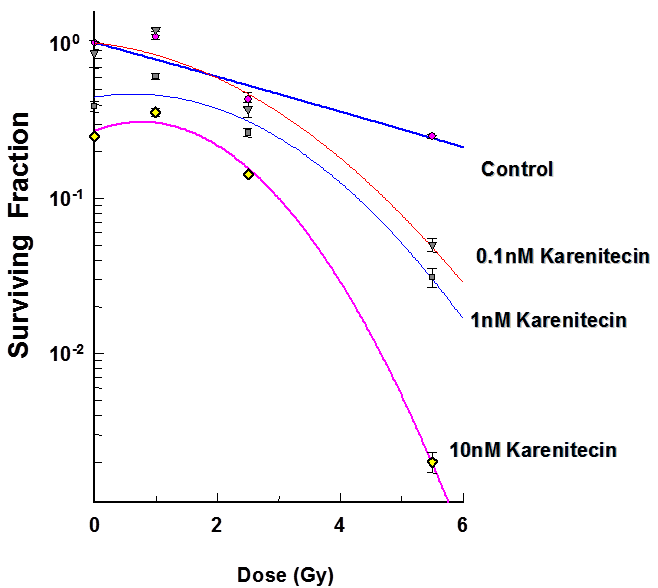
Figure 2: Subconfluent cultures of T98G cells (Figure 1) or MO59K (Figure 2) were treated with 0.1 nM, 1 nM and 10 nM concentrations of karenitecin for 72 hours and subjected to different doses of irradiation (1 Gy – 8.5 Gy). The control set of cells was treated with the indicated doses of radiation, or karenitecin, or with the vehicle control (DMSO) alone. The cells were harvested and an optimum number of cells were plated and allowed to grow for 14 days. The resulting colonies were stained and counted. Each graphed point represents mean values ± SE values of triplicate dishes (variation less than 5%).
Karenitecin induces apoptosis and cell cycle changes in glioma cell lines
Since a number of chemotherapeutic agents eliminate cancer cells by inducing apoptosis, it was of interest if karenitecin induced apoptosis in glioma cell lines. As seen in Figures 3A and 3B, treatment of T98G and MO59K cells with karenitecin revealed a dose and time dependent increase in the sub G0/G1 population of cells indicating the onset of apoptosis. Karenitecin induced cell death was confirmed by dual colored (PI and Annexin V) flow cytometry. Karenitecin treated cells revealed an increase in the early apoptotic [annexin V+/PI¯], apoptotic [annexin V+/PI+] population of cells (Figure 4) indicating the onset of apoptosis
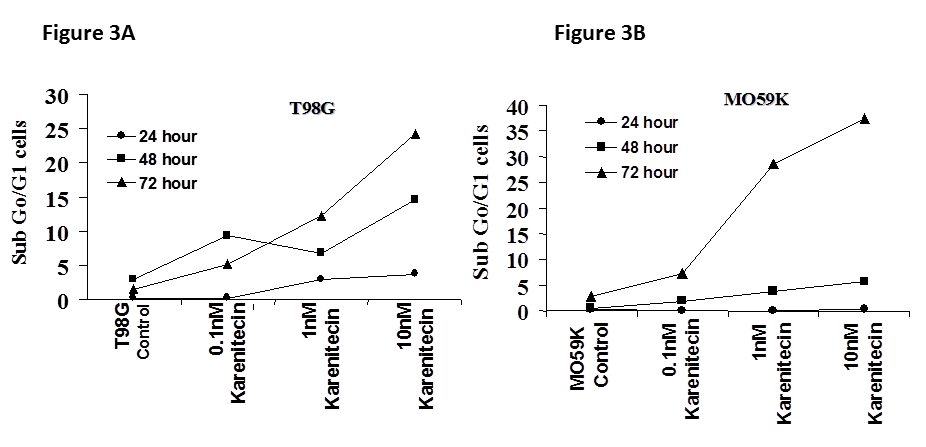
Figures 3A and 3B: The effects of various concentrations of karenitecin on the induction of apoptosis in T98G an4 MO59K cell lines. T98G cells (Figure 3A) or MO59K cells (Figure 3B) were treated with either vehicle DMSO or with indicated concentration of karenitecin. The percent of cells undergoing apoptosis was determined at 24, 48 and 72 hours after treatment by flow cytometry of propidium iodide stained cells. Each point is an average of duplicate dishes (variation <5%).
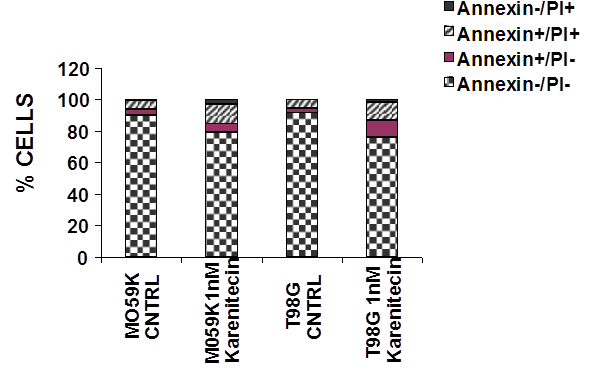
Figure 4: Karenitecin-induced apoptosis of glioma cell lines as determined by dual colored (PI and Annexin V) flow cytometry. The cell lines were treated with or without 1 nM karenitecin for 72hrs. The cells were harvested by cell dissociation solution washed with PBS and subsequently stained with antibody to annexin V conjugated to FITC and with PI [10 µg/ml]. Viable [annexin V¯/PI¯] pre-apoptotic [annexin V+/PI¯], apoptotic [annexin V+/PI+], and the residual damaged [annexin V¯/PI+ cells were quantified by flow cytometric analysis. NOTE: Bar reads top to bottom Annexin¯/PI+, Annexin+/PI+, Annexin+/PI¯, Annexin¯/PI¯
Treatment of T98G and MO59K cells with varying concentrations of karenitecin in the presence and absence of radiation for 72 hours revealed an increase in the G2/M phase of the cell cycle, and a decrease in the G0/G1 phase of the cell cycle (Figures 5 and 6). The increase in the G2/M phase of the cell cycle was dose dependent and also seen in the presence of radiation.
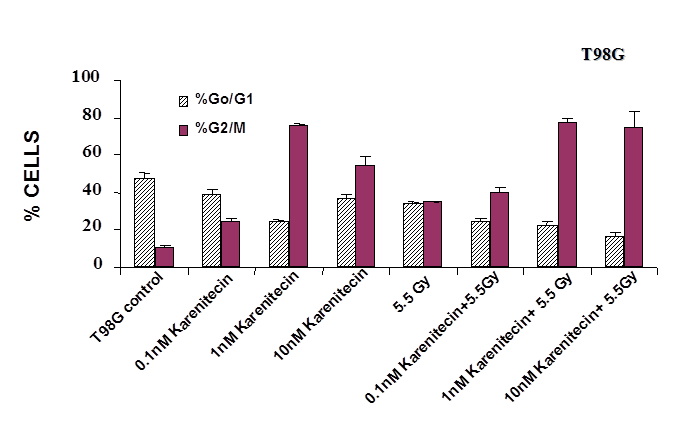
Figure 5: The effects of karenitecin on cell cycle. Subconfluent cultures of T98G cells (Figure 5) or MO59K cells (Figure 6) were treated with either vehicle DMSO or with indicated concentration of karenitecin dissolved in DMSO in the presence and absence of 5.5 Gy radiation for 72 hours. The cells were harvested, fixed in ethanol and stained with propidium iodide. The percent of cells in the G0/G1 and G2/M phases of the cell cycle were determined by flow cytometry. Each graphed point represents mean values ± SE values of triplicate dishes (variation less than 5%).
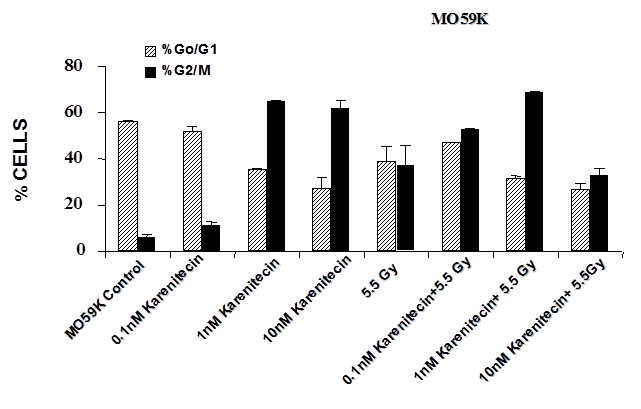
Figure 6: The effects of karenitecin on cell cycle. Subconfluent cultures of T98G cells (Figure 5) or MO59K cells (Figure 6) were treated with either vehicle DMSO or with indicated concentration of karenitecin dissolved in DMSO in the presence and absence of 5.5 Gy radiation for 72 hours. The cells were harvested, fixed in ethanol and stained with propidium iodide. The percent of cells in the G0/G1 and G2/M phases of the cell cycle were determined by flow cytometry. Each graphed point represents mean values ± SE values of triplicate dishes (variation less than 5%).
Karenitecin-treatment increased the levels of cyclins B and D
Distinct cyclins perform different tasks in specific phases of the cell cycle by binding to, and activating different cyclin dependent kinases. Cyclin B promotes G2/M transition, while the D-type cyclins (cyclin D1, D2, and D3) promote cell cycle progression from G1 to S phase39. Since karenitecin induced a block in the G2/M phase of the cell cycle and a decrease in the G0/G1 phase of the cell cycle, we evaluated karenitecin treated glioma cells for the presence of cyclin D1 and B1 in the G0/G1 and G2/M phases of the cell cycle by flow cytometry. The results in Figure 7 revealed an increase in the levels of cyclin B1 predominantly in the G2/M phase of the cell cycle. This result correlated with the increase in the percentage of cells in the G2/M phase of the cell cycle following karenitecin treatment in both the cell lines. There was either no significant change or a slight decrease in the levels of cylcin D in cells present in the G0/G1 phase of the cell cycle (Figure 8). This finding correlated with the decrease in the percentage of cells in the G0/G1 phase of the cell cycle. A slight increase in the expression of cyclin B1 was seen in the G0/G1 phase of the cell cycle and an increase in the expression of cyclin D was also observed in the G2/M phase of the cell cycle, which could be attributed to the unscheduled cyclin production in glioma cells (Figure 8).
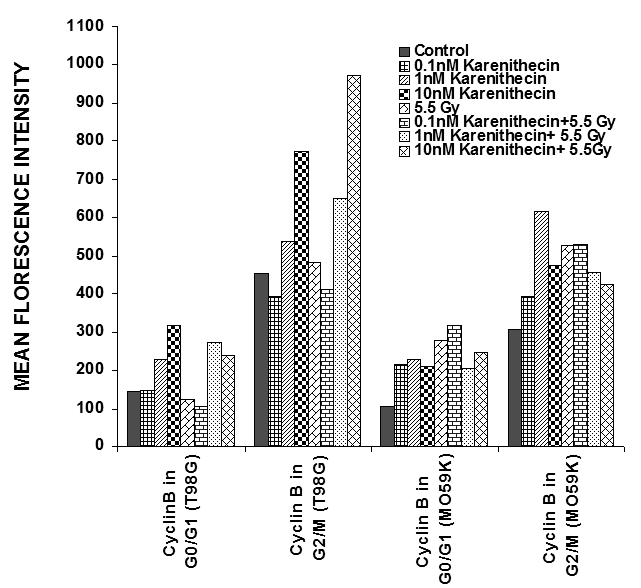
Figure 7: Bivariate analysis of DNA content and cyclins following karenitecin treatment: Subconfluent cultures of T98G and MO59K cells were treated with the indicated concentration of karenitecin in the presence and absence of radiation for 72 hours. Following treatment, the cells were harvested, fixed in ethanol, and permeabilized. The cells were stained using specific antibodies for cyclin D1 or cyclin B1 or the relevant isotype control and with goat anti-mouse FITC. The cells were stained with PI solution and analyzed on a FACScan flow cytometer (Becton Dickinson, San Jose, CA). PI staining was used to view different phases of the cell cycle and the cyclin staining in the G0/G1 and G2/M phase of the cell cycle was estimated using the cell quest program. The graph represents the MFI recorded for cyclin B1 and cyclin D1 staining after subtracting the MFI of the isotype control of each treated sample. Figure 7 reveals the effect of karenitecin on cyclin B1 levels, while Figure 8 reveals the effect of karenitecin on the levels of cyclin D1 in T98G and MO59K cells in the G0/G1and G2/M phase of the cell cycle.
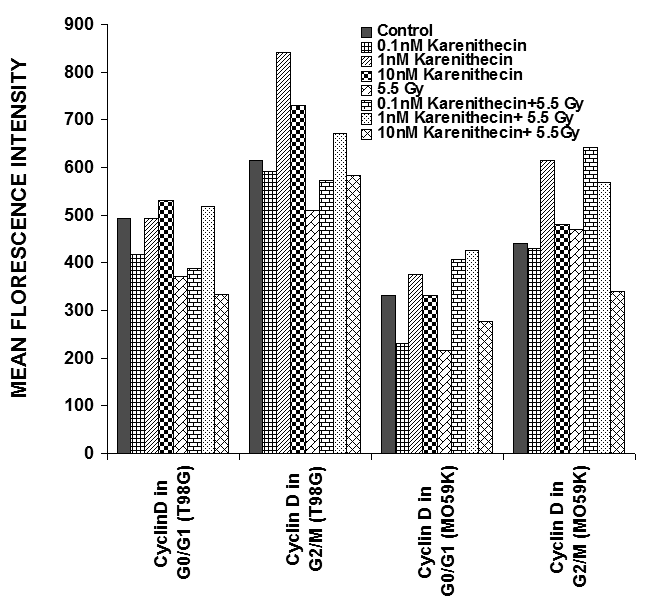
Figure 8: Bivariate analysis of DNA content and cyclins following karenitecin treatment: Subconfluent cultures of T98G and MO59K cells were treated with the indicated concentration of karenitecin in the presence and absence of radiation for 72 hours. Following treatment, the cells were harvested, fixed in ethanol, and permeabilized. The cells were stained using specific antibodies for cyclin D1 or cyclin B1 or the relevant isotype control and with goat anti-mouse FITC. The cells were stained with PI solution and analyzed on a FACScan flow cytometer (Becton Dickinson, San Jose, CA). PI staining was used to view different phases of the cell cycle and the cyclin staining in the G0/G1 and G2/M phase of the cell cycle was estimated using the cell quest program. The graph represents the MFI recorded for cyclin B1 and cyclin D1 staining after subtracting the MFI of the isotype control of each treated sample. Figure 7 reveals the effect of karenitecin on cyclin B1 levels, while Figure 8 reveals the effect of karenitecin on the levels of cyclin D1 in T98G and MO59K cells in the G0/G1and G2/M phase of the cell cycle.
Flavopirdol induced radiosensitization in T98G cells
Since the onset of apoptosis by karenitecin was accompanied by an arrest in the GG2/M phase of the cell cycles, it was of interest to study the combined effects of karenitecin and novel inhibitors of cyclin dependent kinases (CDK).
T98G cells were treated with varying concentrations of flavopridol at concentrations of 50 nM - 200 nM for 72 hours the cell survival was determined by the clonogenic survival assay. The results showed that flavopridol did not drastically affect cell survival (Figure 9) but higher concentrations between 300-500 nM resulted in reduction of the surviving fraction in these cell lines. Radiosensitization experiments in the dose range between 50 – 200 nM of flavopridol. Pretreatment of T98G cells (Figure 9) with flavopridol revealed a dose dependent sensitization to radiation induced cell death.
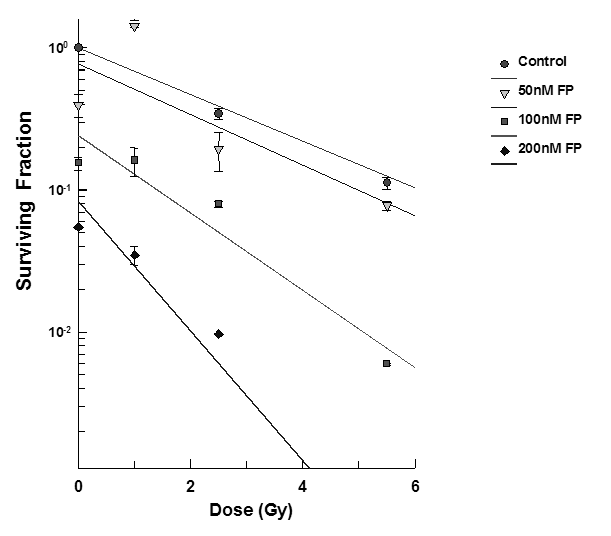
Figure 9: Subconfluent cultures of T98G cells were treated with 50 – 200 nM concentrations of flavopridol for 72 hours and subjected to different doses of irradiation as shown. The control set of cells was treated with the indicated doses of radiation, or flavopridol, or with medium alone. The cells were harvested and an optimum number of cells were plated and allowed to grow for 14 days. The resulting colonies were stained and counted. Each graphed point represents mean values ± SE values of triplicate dishes (variation less than 5%). Note: Control bar followed by increasing doses of karenitecin seen right to left.
In order to determine the time of pretreatment needed for the radiosensitization to occur, T98G cells were treated with 200 nM of flavopridol between 2-72 hours. At the end of each pretreatment the cells were harvested and the surviving fraction was determined. The results showed that exposure to flavopridol for 8 hours was sufficient to cause radiosensitization (Figure 10).
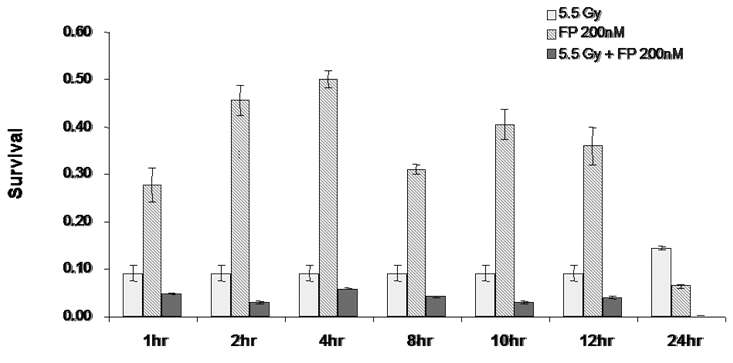
Figure 10: Time course for flavopridol mediated radiosensitization: Subconfluent cultures of T98G cells were treated with 200 nM flavopridol for 1-24 hours and subsequently to 5.5 Gy of radiation. Control cells were treated with the indicated doses of radiation alone. The cells were harvested and an optimum number of cells were allowed to grow for 14 days. The resulting colonies were stained with crystal violet and counted. Survival was determined as the ratio of plating efficiencies for each treated group to that of the un-irradiated control. Each graphed point represents mean values ± SE values of triplicate dishes.
Flavopridol induced apoptosis accompanied changes in cell cycle progression
The next set of experiments was designed to determine the mode of death induced by flavopridol. Sub-confluent cultures of T98G cells were treated with flavopridol (50 nM – 200 nM) for a period of 24, 48 and 72 hours and percentages of cells undergoing apoptosis and the changes in different phases of the cell cycle were quantitated by flow cytometry. The results revealed a dose dependent increase in the sub G0/G1 population of the cells with time indicating that cells could be dying by apoptosis. Treatment with flavopridol caused a predominant increase in the G2/M phase of the cell cycle, and a decrease in the G0/G1 phase of the cell cycle. The increase in the G2/M phase of the cell cycle occurred with 100 nM of flavopridol at 24 hours. The G2/M block was sustained up to 72 hours of drug treatment, indicating that cells treated with radiation were predominantly in the G2/M phase of cell cycle (Figure 11).
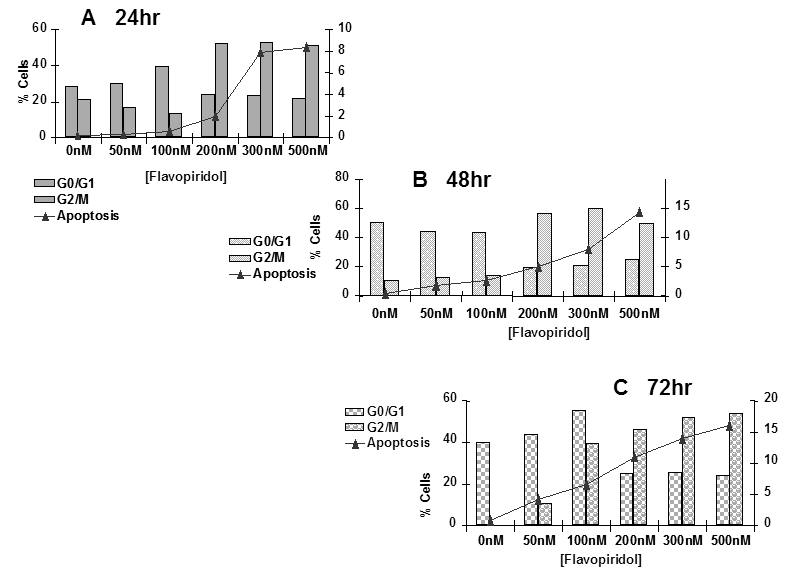
Figure 11: Cell cycle analysis of flavopridol treated T98G cells. Subconfluent cultures of T98G cells were treated with 50 – 500 nM of flavopridol for 24, 48 or 72 hrs (A, B, C respectively). The cells were harvested, fixed in ice cold ethanol and stained using PI (20 ug/ml). The percent of cells in the G0/G1, G2/M phases of the cell cycle and the percentage of cells undergoing apoptosis (Sub G0/G1) was determined by flow cytometric analysis. Each point is an average of two separate experiments (variation <5%).
Karenitecin and flavopridol pretreatment augmented radiation induced cell death
T98G cells were treated with a combination of flavopridol [50 nM and100 nM] and karenitecin [0.1 nM and 1 nM] for 72 hours. The doses of flavopridol and karenitecin and radiation were clinically relevant. 72 hours was chosen as it caused the accumulation of cells in the G2/M phase of the cell cycle. A combination of karenitecin and flavopridol increased cell death greater than the additive effects caused by each of the drugs alone. This effect was further enhanced in the presence of radiation (Figure 12). Both flavopridol and karenitecin alone or in combination caused an increase in the G2/M phase of the cell cycle (Figure 13). These results demonstrate the ability of karenitecin and flavopridol to induce radiosensitization in glioma cells and indicate the ability of karenitecin to synergize with CDK inhibitors.
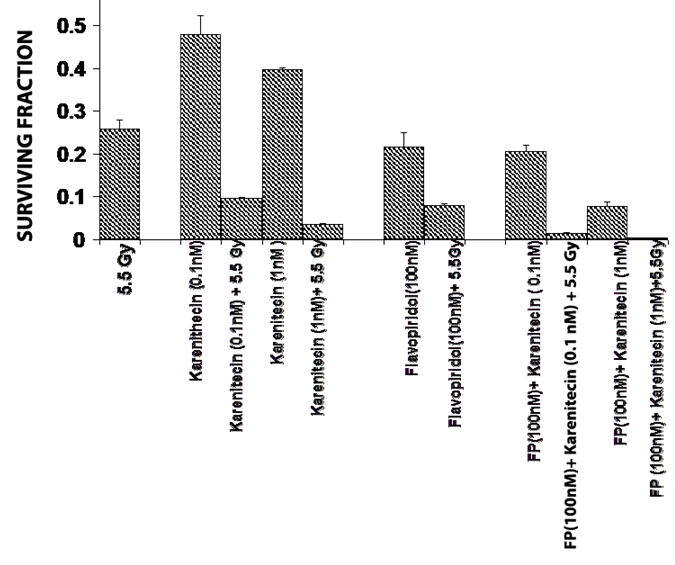
Figure 12: Combined effects of karenitecin and flavopridol: Subconfluent cultures of the T98G cells were treated with the indicated concentrations of flavopridol, or karenitecin, or a combined treatment of both the drugs 72 hours followed by treatment with 5.5 Gy of radiation. Control set of cells was treated with 5.5 Gy radiation, or treated with DMSO alone. The cells were harvested and an optimum number of cells were plated and allowed to grow for 14 days. The resulting colonies were stained and counted. Each graphed point represents mean values ± SE values of triplicate dishes.
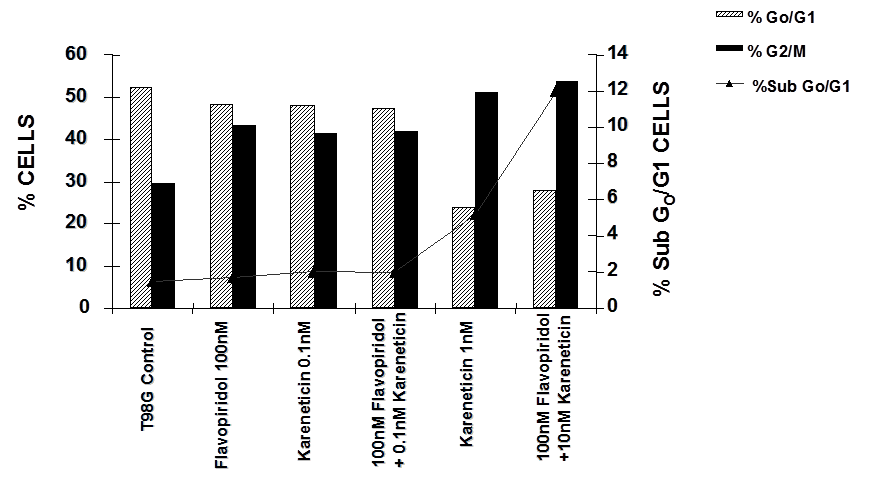
Figure 13: The effect of karenitecin and flavopridol on cell cycle and induction of apoptosis in T98G cell line. Subconfluent cells of T98G cells were treated with either vehicle DMSO or with indicated concentration of karenitecin and flavopridol dissolved in DMSO. The percent of cells undergoing apoptosis and in the G0/G1 and G2/M phases of the cell cycle was determined at 72 hours after treatment by flow cytometry of propidium iodide stained cells. Each point is an average of duplicate dishes (variation <5%).
Discussion
Knowledge concerning the regulatory molecules involved in the control of growth and malignant progression is an essential component to conceptualization of effective agents for treatment of malignant glioma. The efficacy of many antitumor agents is dependent on DNA replication and cell cycle progression.
The present study evaluates the usefulness the topisomerase I inhibitor karenitecin and the cyclin dependent kinase inhibitor flavopridol as potential radiosensitizers in malignant glioma. Multiple laboratories have shown flavopridol to induce apoptosis independent of the p53 and Rb status of the cell, promote differentiation, inhibit angiogenesis, modulate transcriptional events and block cell cycle progression via a transient G2/M block in the cell cycle.
We have shown that pretreatment of glioma cell lines with karenitecin induced radiosensitization at sub-toxic concentrations of karenitecin and in the clinically relevant dose range of radiation. Karenitecin induced apoptosis, a block in the G2/M phase of the cell cycle and enhanced the expression of cyclin B1. Our results are in agreement with the reports of Yin et al28, where the authors demonstrated the up-regulation of cyclin B/cdc2 protein expression and an increase in the phosphorylation of the G2/M checkpoint kinase chk1 in colon cancer cell line HCT-8 by karenitecin. Our results also reveal an increase in the cyclin D levels in glioma cells treated with both karenitecin and radiation. Over expression of cyclin D1 has been reported to alter sensitivity toward ionizing radiation in breast cancer cells40,41; this could explain the increased levels of cyclin D in cells treated with both karenitecin and radiation. Karenitecin has been shown to induce apoptosis in head and neck cancer cell line A253 and cause growth inhibition of xenografts derived from childhood and adult high-grade glioma, medulloblastomas, ependymomas, as well as sub-lines resistant to procarbazine and busulfan by karenitecin28,29. This is the first report of the karenitecin as a radiosensitizer in glioma cells at clinically relevant doses of karenitecin and radiation.
Exposure to DNA damaging agents leads to growth arrest in the G1 and G2 phases of the cell cycle thereby preventing the cells from replicating and separating the damaged DNA. The growth arrest is due to activation of different cell cycle checkpoints surveillance proteins, which are activated by DNA damage or aberrant DNA replication. It is generally believed that the cell cycle arrest provides the cells with additional time to repair the potentially lethal DNA lesions, thereby rendering them more resistant to the cytotoxic effects of DNA damaging agents. Abrogation of the G2 checkpoint is accompanied by increased cytotoxicity of many anticancer agents both in vitro and in vivo. The cyclin dependent kinase 2, cyclin B1 complex plays a key role in regulating the G2/M phase of the cell cycle. Although activation of the cyclin-dependent kinase CDC2 mediates G2/M transition in all tumor cells studied to date, regulation of CDC2 varies between tumor types. Persistent hyper phosphorylation of kinase and reduced cyclin expression have been implicated as mediators of treatment-induced G2 delay in different tumor models.
Flavopridol is a potent inhibitor of most CDKs, including CDK1, CDK2, CDK4, and CDK79,23,42,43. Multiple laboratories have shown flavopridol to induce apoptosis independent of the p53 and Rb status of the cell, promote differentiation, inhibit angiogenesis, modulate transcriptional events and block cell cycle progression via a transient G2/M block in the cell cycle. There have been reports on the synergistic effects of flavopridol in combination with other therapeutic drugs such as mitomycin, cytarabine, paclitaxel, and cisplatin44-46.
We have demonstrated that flavopridol could be a radiosensitizer at clinically relevant doses. Flavopridol induced apoptosis and a G2/M block in T98G cells. Flavopridol induced cell death was confirmed by dual colored (PI and Annexin V) flow cytometry (data not shown). Alonso et al47 reported the ability of flavopridol to induce apoptosis in a panel of glioma cell lines via a caspase-independent mechanism. They also reported the downregualtion of MDM2 mRNA and protein levels and cyclin D1 levels independent of Rb or p53 tumor suppressor pathway alterations. MDM2 protein regulates Rb and p53 function and under certain circumstances confers oncogenic properties48. Loss of MDM2 expression in tumor cells by flavopridol treatment could promote increased radiosensitivity.
Both kareniticin and flavopiridiol induce an arrest in the G2/M phase of the cell cycle. Their combined action affects cell replication and cell cycle regulation. Cells harboring mutated p53 (T98G) display a block in the G2/M phase of the cell cycle in response to Ionizing radiation indicating that the G2-phase block is a crucial event in the damage response of both these drugs and radiation. This G2-phase block is a crucial event in the damage response that can be manipulated to achieve a significant enhancement of drug toxicity potentially.
DNA might be a second target of flavopridol binding, as well as for karenitecin, and their combined effects could be also cytotoxic to non-cycling cancer cells.
In summary, we have shown, that orally administrable topoisomerase inhibitor karenitecin (BNP1350) and the cyclin dependent kinase inhibitor flavopridol both induce apoptosis, and radiosensitization of glioma cell lines. The radiosensitization appears to be independent of the p53 status since T98G cells have been demonstrated to harbor mutated p53. The combined effects of karenitecin and radiation or flavopridol with radiation or a combination of both the drugs with ionizing radiation could cause the shortening of the G2/M delay and cause sensitization of glioma cells to cytotoxic treatment.
Future studies aimed at understanding the underlying mechanisms of molecular radiosensitization with karenitecin and or flavopridol could provide valuable information for its further preclinical or clinical evaluation either as single agent or in combination with other agents in the treatment of malignant glioma.
Acknowledgement
Funded in part by: Kathleen Reader Neuro-Oncology Fund; Human Oncology Neuro-Oncology Fund; P30 CA014520 (Core Grant, University of Wisconsin Carbone Cancer Center.
We thank Dr. Frederick H. Hausheer, Bionumerik Pharmaceuticals Inc., (San Antonio TX); for providing us karenitecin used in the present study. Flow cytometry provided by the University of Wisconsin Carbone Cancer Center grant P30 CA014520. We also thank Dr. Mark Ritter (University of Wisconsin Madison Comprehensive Cancer Center) for providing us flavopridol used in the present study.
References
- Bible KC, Bible RH, Jr., Kottke TJ, Svingen PA, Xu K, Pang YP, et al. Flavopiridol binds to duplex DNA. Cancer Res 2000;60: 2419-28.
- Bible KC, Kaufmann SH. Flavopiridol: a cytotoxic flavone that induces cell death in noncycling A549 human lung carcinoma cells. Cancer Res 1996;56: 4856-61.
- Innocenti F, Stadler WM, Iyer L, Ramirez J, Vokes EE, Ratain MJ. Flavopiridol metabolism in cancer patients is associated with the occurrence of diarrhea. Clin Cancer Res 2000;6: 3400-5.
- Monga M, Sausville EA. Developmental therapeutics program at the NCI: molecular target and drug discovery process. Leukemia 2002;16: 520-6.
- Sausville EA, Zaharevitz D, Gussio R, Meijer L, Louarn-Leost M, Kunick C, et al. Cyclin-dependent kinases: initial approaches to exploit a novel therapeutic target. Pharmacol Ther 1999;82: 285-92.
- Senderowicz AM. Cyclin-dependent kinases as targets for cancer therapy. Cancer Chemother Biol Response Modif 2002;20: 169-96.
- Shapiro GI, Supko JG, Patterson A, Lynch C, Lucca J, Zacarola PF, et al. A phase II trial of the cyclin-dependent kinase inhibitor flavopiridol in patients with previously untreated stage IV non-small cell lung cancer. Clin Cancer Res 2001;7: 1590-9.
- Stadler WM, Vogelzang NJ, Amato R, Sosman J, Taber D, Liebowitz D, et al. Flavopiridol, a novel cyclin-dependent kinase inhibitor, in metastatic renal cancer: a University of Chicago Phase II Consortium study. J Clin Oncol 2000;18: 371-5.
- Wang HK. The therapeutic potential of flavonoids. Expert Opin Investig Drugs 2000;9: 2103-19.
- Drewinko B, Patchen M, Yang LY, Barlogie B. Differential killing efficacy of twenty antitumor drugs on proliferating and nonproliferating human tumor cells. Cancer Res 1981;41: 2328-33.
- Cheng M, Olivier P, Diehl JA, Fero M, Roussel MF, Roberts JM, et al. The p21(Cip1) and p27(Kip1) CDK 'inhibitors' are essential activators of cyclin D-dependent kinases in murine fibroblasts. EMBO J 1999;18: 1571-83.
- Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev 1999;13: 1501-12.
- Johnson DG, Walker CL. Cyclins and cell cycle checkpoints. Annu Rev Pharmacol Toxicol 1999;39: 295-312.
- Huschtscha LI, Reddel RR. p16(INK4a) and the control of cellular proliferative life span. Carcinogenesis 1999;20: 921-6.
- Soos TJ, Park M, Kiyokawa H, Koff A. Regulation of the cell cycle by CDK inhibitors. Results Probl Cell Differ 1998;22: 111-31.
- Chien M, Astumian M, Liebowitz D, Rinker-Schaeffer C, Stadler WM. In vitro evaluation of flavopiridol, a novel cell cycle inhibitor, in bladder cancer. Cancer Chemother Pharmacol 1999;44: 81-7.
- Kelland LR. Flavopiridol, the first cyclin-dependent kinase inhibitor to enter the clinic: current status. Expert Opin Investig Drugs. 2000;9: 2903-11.
- Schwartz GK, Ilson D, Saltz L, O'Reilly E, Tong W, Maslak P, et al. Phase II study of the cyclin-dependent kinase inhibitor flavopiridol administered to patients with advanced gastric carcinoma. J Clin Oncol. 2001;19: 1985-92.
- Schwartz GK, O'Reilly E, Ilson D, Saltz L, Sharma S, Tong W, et al. Phase I study of the cyclin-dependent kinase inhibitor flavopiridol in combination with paclitaxel in patients with advanced solid tumors. J Clin Oncol. 2002;20: 2157-70.
- Tan AR, Headlee D, Messmann R, Sausville EA, Arbuck SG, Murgo AJ, et al. Phase I clinical and pharmacokinetic study of flavopiridol administered as a daily 1-hour infusion in patients with advanced neoplasms. J Clin Oncol. 2002;20: 4074-82.
- Zhai S, Sausville EA, Senderowicz AM, Ando Y, Headlee D, Messmann RA, Arbuck S, Murgo AJ, Melillo G, Fuse E, Figg WD. Clinical pharmacology and pharmacogenetics of flavopiridol 1-h i.v. infusion in patients with refractory neoplasms. Anticancer Drugs. 2003;14: 125-35.
- Jager W, Zembsch B, Wolschann P, Pittenauer E, Senderowicz AM, Sausville EA, Sedlacek HH, Graf J, Thalhammer T. Metabolism of the anticancer drug flavopiridol, a new inhibitor of cyclin dependent kinases, in rat liver. Life Sci. 1998;62: 1861-73.
- Sedlacek HH. Mechanisms of action of flavopiridol. Crit Rev Oncol Hematol. 2001;38: 139-70.
- Motwani M, Li X, Schwartz GK. Flavopiridol, a cyclin-dependent kinase inhibitor, prevents spindle inhibitor-induced endoreduplication in human cancer cells. Clin Cancer Res 2000;6: 924-32.
- Owa T, Yoshino H, Yoshimatsu K, Nagasu T. Cell cycle regulation in the G1 phase: a promising target for the development of new chemotherapeutic anticancer agents. Curr Med Chem. 2001;8: 1487-503.
- Pollack IF, Erff M, Bom D, Burke TG, Strode JT, Curran DP. Potent topoisomerase I inhibition by novel silatecans eliminates glioma proliferation in vitro and in vivo. Cancer Res. 1999;59: 4898-905.
- Matsui S, Endo W, Wrzosek C, Haridas K, Seetharamulu P, Hausheer FH, et al. Characterisation of a synergistic interaction between a thymidylate synthase inhibitor, ZD1694, and a novel lipophilic topoisomerase I inhibitor karenitecin, BNP1100: mechanisms and clinical implications. Eur J Cancer. 1999;35: 984-93.
- Yin MB, Guo B, Vanhoefer U, Azrak RG, Minderman H, Frank C, et al. Characterization of protein kinase chk1 essential for the cell cycle checkpoint after exposure of human head and neck carcinoma A253 cells to a novel topoisomerase I inhibitor BNP1350. Mol Pharmacol. 2000;57: 453-9.
- Keir ST, Hausheer F, Lawless AA, Bigner DD, Friedman HS. Therapeutic activity of 7-[(2-trimethylsilyl)ethyl)]-20 (S)-camptothecin against central nervous system tumor-derived xenografts in athymic mice. Cancer Chemother Pharmacol. 2001;48: 83-7.
- Smith JA, Hausheer F, Newman RA, Madden TL. Development of a high-performance liquid chromatographic method to determine the concentration of karenitecin, a novel highly lipophilic camptothecin derivative, in human plasma and urine. J Chromatogr B Biomed Sci Appl. 2001;759: 117-24.
- Grossman SA, Carson KA, Phuphanich S, Batchelor T, Peereboom D, Nabors LB, et al. New Approaches to Brain Tumor Therapy CNSC. Phase I and pharmacokinetic study of karenitecin in patients with recurrent malignant gliomas. Neuro Oncol. 2008;10: 608-16.
- Rubenstein M, Shaw M, Mirochnik Y, Slobodskoy L, Glick R, Lichtor T, et al. In vivo establishment of T98G human glioblastoma. Methods Find Exp Clin Pharmacol. 1999;21: 391-3.
- Stein GH. T98G: an anchorage-independent human tumor cell line that exhibits stationary phase G1 arrest in vitro. J Cell Physiol. 1979;99: 43-54.
- Wang J, Hu L, Allalunis-Turner MJ, Day RS, 3rd, Deen DF. Radiation-induced damage in two human glioma cell lines as measured by the nucleoid assay. Anticancer Res. 1997;17: 4615-8.
- Darzynkiewicz Z, Li X, Gong J. Assays of cell viability: discrimination of cells dying by apoptosis. Methods Cell Biol. 1994;41: 15-38.
- Gorczyca W, Melamed MR, Darzynkiewicz Z. Analysis of apoptosis by flow cytometry. Methods Mol Biol. 1998;91: 217-38.
- Del Bino G, Darzynkiewicz Z, Degraef C, Mosselmans R, Fokan D, Galand P. Comparison of methods based on annexin-V binding, DNA content or TUNEL for evaluating cell death in HL-60 and adherent MCF-7 cells. Cell Prolif. 1999;32: 25-37.
- Darzynkiewicz Z, Traganos F. Measurement of apoptosis. Adv Biochem Eng Biotechnol. 1998;62: 33-73.
- Grana X, Reddy EP. Cell cycle control in mammalian cells: role of cyclins, cyclin dependent kinases (CDKs), growth suppressor genes and cyclin-dependent kinase inhibitors (CKIs). Oncogene. 1995;11: 211-9.
- Coco Martin JM, Balkenende A, Verschoor T, Lallemand F, Michalides R. Cyclin D1 overexpression enhances radiation-induced apoptosis and radiosensitivity in a breast tumor cell line. Cancer Res. 1999;59: 1134-40.
- Turner BC, Gumbs AA, Carter D, Glazer PM, Haffty BG. Cyclin D1 expression and early breast cancer recurrence following lumpectomy and radiation. Int J Radiat Oncol Biol Phys. 2000;47: 1169-76.
- Carlson BA, Dubay MM, Sausville EA, Brizuela L, Worland PJ. Flavopiridol induces G1 arrest with inhibition of cyclin-dependent kinase (CDK) 2 and CDK4 in human breast carcinoma cells. Cancer Res. 1996;56: 2973-8.
- Kim SH, Schulze-Gahmen U, Brandsen J, de Azevedo Junior WF. Structural basis for chemical inhibition of CDK2. Prog Cell Cycle Res. 1996;2: 137-45.
- Bible KC, Kaufmann SH. Cytotoxic synergy between flavopiridol (NSC 649890, L86-8275) and various antineoplastic agents: the importance of sequence of administration. Cancer Res. 1997;57: 3375-80.
- Motwani M, Delohery TM, Schwartz GK. Sequential dependent enhancement of caspase activation and apoptosis by flavopiridol on paclitaxel-treated human gastric and breast cancer cells. Clin Cancer Res. 1999;5: 1876-83.
- Schwartz GK, Farsi K, Maslak P, Kelsen DP, Spriggs D. Potentiation of apoptosis by flavopiridol in mitomycin-C-treated gastric and breast cancer cells. Clin Cancer Res. 1997;3: 1467-72.
- Alonso M, Tamasdan C, Miller DC, Newcomb EW. Flavopiridol induces apoptosis in glioma cell lines independent of retinoblastoma and p53 tumor suppressor pathway alterations by a caspase-independent pathway. Mol Cancer Ther. 2003;2: 139-50.
- Berger M, Vogt Sionov R, Levine AJ, Haupt Y. A role for the polyproline domain of p53 in its regulation by Mdm2. J Biol Chem. 2001;276: 3785-90.