
Lung inflammation regulates autoimmunity in the central nervous system
Masashi Kanayama1, Mari L. Shinohara2*
Abstract
Abstract Autoimmune diseases of the central nervous system (CNS), such as multiple sclerosis (MS), are characterized by infiltration of pathogenic immune cells in the CNS. Cells infiltrated in the CNS express pro-inflammatory molecules and cause demyelination. The direct impact of lung immune responses was not previously considered in the pathogenesis of CNS inflammation. However, it recently became clear that the lung acts as a hub organ of pathogenic T cells that are migrating to the CNS. More recent studies further showed that inflamed lung has a critical impact on autoimmune CNS inflammation. Here, we discuss the contribution of the lung in the pathogenesis of CNS autoimmune diseases based on recent reports.
Addreviations
BBB: blood brain barrier; CNS: central nervous system; CKO: conditional knockout; EAE: experimental autoimmune encephalomyelitis; LAP: LC3-associated phagocytosis; MS: multiple sclerosis.
Background
The central nervous system (CNS), including the brain and the spinal cord, is a vital organ to maintain our life. To maintain its integrity by evacuating from influences of peripheral immune responses, the CNS is segregated by the blood brain barrier (BBB). However, immune cells, especially autoreactive T cells, invade into the CNS by exceeding BBB and cause inflammation and tissue damages during autoimmune CNS inflammation. Such events are seen in multiple sclerosis (MS) and its animal model experimental autoimmune encephalomyelitis (EAE). Thus, infiltration of peripheral immune cells into the CNS is a critical event, disturbing immune homeostasis in the CNS. Because MS and EAE are T cell-dependent autoimmune diseases, infiltration of T cells is considered to be essential to develop the diseases. In particular, Th17 cells, an encephalitogenic helper T cell subset producing a cytokine IL-17, are known to play a critical role in the pathogenesis of MS and EAE1,2. Therefore, multiple therapeutic approaches, which target the mobility or functions of T cells including Th17 cells, have been approved by Food and Drug Administration or studied on clinical trial3.
The lung is a respiratory organ, which converts blood carbon dioxide to oxygen. Circulating blood passes through the lung before circulating through the body. Since the lung is at the center of the circulation system, its strong influence on blood cells is expected. A seminal article by Odoardi et al. reported that pathogenic T cells are licensed in the lung to migrate into the CNS during EAE4. The licensing in the lung was demonstrated as enhanced expression of genes encoding chemokine receptors and integrins to promote T cell migration4. After T cells’ temporary stay in the lung, licensed T cells re-enter the blood stream, then migrate to the CNS. In contrast, T cells that have not licensed in the lungs failed to migrate to the CNS4. Although the detailed molecular mechanism of T cell licensing in the lung remains largely unknown, the lung clearly plays a critical role in the development of CNS autoimmunity. In the next section, we discuss impacts of lung inflammation during EAE development by reviewing two recent articles.
Inflammation and infection in the lungs affect the development of EAE
The lungs are constantly exposed to environmental stimuli such as pathogens, air dust, and oxidative stress. To maintain the immune homeostasis, the lung has a unique immune system to avoid overreacting to the environmental stimuli5,6. Recent reports from another group and us demonstrated that the course of EAE development is altered when the lung immune homeostasis is disturbed by inflammation or infections7,8. Below, we discuss the two cases of scenario in EAE.
Case 1 -- Spontaneous lung inflammation protecting mice from EAE
We found that the lung-specific inflammation caused delayed EAE onset7. The finding was seen in mice lacking ATG7 in myeloid cells. Since ATG7 (Autophagy Related 7) is an essential protein for autophagy and LC3-associated phagocytosis (LAP)9,10, ATG7-deficient cells do not perform autophagy and LAP. Myeloid cell-specific ATG7 (Atg7 CKO mice) conditional knockout mice develop spontaneous pulmonary inflammation due to the heightened sensitivity to environmental microbes11,12. However, importantly, inflammation in the mice is subclinical and restricted in the lungs11. The lungs of the Atg7 CKO mice is characterized by enhanced expression of pro-inflammatory cytokines and chemokines, in addition to the infiltration of innate immune cells11.
Delayed EAE onset is found in the Atg7 CKO mice11. Interestingly, lungs of the Atg7 CKO mice stall pathogenic T cells, Th17 cells in particular, on the way to migrate to the CNS (Fig. 1). This causes the delay of EAE onset, although the mice eventually develop severe EAE as WT mice do11. It should be noted that the delayed EAE onset is not due to defects in pathogenic T cell development because Atg7 CKO mice have normal numbers of effector T cells and T cell expansion in the draining lymph nodes. EAE onset is also delayed when pathogenic WT Th17 cells were adoptively transferred in Atg7 CKO recipients11, suggesting that the delayed EAE onset is not caused by T cells. It is possible that elevated CCL20 levels in the inflamed lungs are involved, at least in part. CCL20 is a chemokine to attracts Th17 cells through its receptor, CCR613. Lungs in Atg7 CKO mice have enhanced Ccl20 gene expression11, which may explain the stalling of Th17 cells in the lung on the way to the CNS. Indeed, CCL20 is expressed and attracts Th17 cells in the entrance points for inflammatory cells into the CNS, the choroid plexus of the brain and the 5th lumber of the spinal cord14-16. Ccl20 expression is induced by intratracheal instillation of a low dosage of LPS, and inflammation in the lung can be induced11. The LPS instillation delays EAE onset due to the accumulation of Th17 cells in the lung11. Thus, lung inflammation stalls pathogenic T cell migration en route to the CNS during EAE development (Fig. 1).
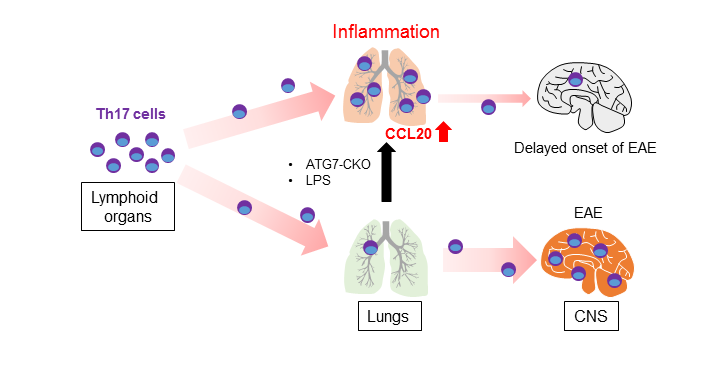
Figure 1: Stalling Th17 cells in the inflamed lung during EAE development.
Case 2 – Bacterial infection in the lung attenuating EAE progression
In the second article, Edwards et al. reported attenuated EAE severity with aerosol-inoculated respiratory bacterial infection with Bordetella pertussis8. Although the study did not mention the possible accumulation of pathogenic cells in the lung, the data showed significantly decreased infiltration of Th17 cells, neutrophils, and macrophages in the brain. The study suggests that the attenuation of EAE is due to IL-10-mediated suppression of T cell licensing in the lung, as well as downregulation of VLA-4 and LFA-1 expression on CD4+ T cells8. Therefore, the data also suggested the attenuation of the T cell-intrinsic ability to migrate to the CNS.
The two recent studies discussed above on EAE with lung inflammation or pulmonary infection did not show the same outcomes, particularly in disease severity. However, the studies revealed the pathogenic conditions in the lung have a significant impact on the development of the CNS autoimmune disease, and it will be of future interest to clarify what type of pulmonary inflammation is protective.
Impact of lung immune responses in pathogenesis of MS
Since we have discussed two reports on EAE, this section focuses on lung immunity and MS. Some reports suggested the connection between the lung and progression of human CNS inflammation in MS. For example, cigarette smoking has been known to increase a risk of MS17,18 and to decrease Ccl20 expression in the lungs of human19. Together with our EAE results7, these human studies may implicate the decrease of Ccl20 expression could shorten the period for Th17 cells to stay in the lung of smokers. At the same time, the period may be short but sufficient to license T cells to be encephalitogenic. In addition, upper respiratory infection with viruses is also a risk factor to trigger MS symptom in relapsing-remitting MS patients20-22. In this case, virus infection is considered to trigger pathogenic Th1 responses in MS, as well as possible epitope spreading to trigger MS. That said, it does not necessarily mean that lung inflammation could delay or attenuate CNS autoimmunity as seen in EAE, but inflammation near the lung could simply trigger and enhance immune responses.
Concluding remarks
As we discussed in this review, the lung plays an important role as a hub of pathogenic T cells to migrate towards the CNS. The mechanism of T cell licensing in the lungs has not fully understood yet, and inflammation in the lung and proximal tissues could either ameliorate or exacerbates CNS autoimmune diseases. However, if the mechanism becomes clear, it might be possible to manipulate pathogenic T cell migration to the CNS through the lung. Treatment by drug inhalation from the mouth is not invasive and an effective route to modify immune responses in the lung. Controlling CNS autoimmune diseases, such as MS, by modifying the lung immune system may be considered to be a new approach to treat the diseases
References
- Kebir H, Kreymborg K, Ifergan I, Dodelet-Devillers A, Cayrol R, Bernard M, et al. Human TH17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat Med. 2007; 13: 1173-1175.
- Sweeney CM, Lonergan R, Basdeo SA, Kinsella K, Dungan LS, Higgins SC, et al. IL-27 mediates the response to IFN-beta therapy in multiple sclerosis patients by inhibiting Th17 cells. Brain Behav Immun. 2011; 25: 1170-1181
- Eckstein C, Bhatti MT, Currently approved and emerging oral therapies in multiple sclerosis: An update for the ophthalmologist. Surv Ophthalmol. 2016; 61: 318-332.
- Odoardi F, Sie C, Streyl K, Ulaganathan VK, Schläger C, Lodygin D, et al. T cells become licensed in the lung to enter the central nervous system. Nature. 2012; 488: 675-679.
- Hussell T, Bell TJ, Alveolar macrophages: plasticity in a tissue-specific context. Nat Rev Immunol. 2014; 14: 81-93.
- Soroosh P, Doherty TA, Duan W, Mehta AK, Choi H, Adams YF, et al. Lung-resident tissue macrophages generate Foxp3+ regulatory T cells and promote airway tolerance. J Exp Med. 2013; 210: 775-788.
- Kanayama M, Danzaki K, He YW, Shinohara ML. Lung inflammation stalls Th17-cell migration en route to the central nervous system during the development of experimental autoimmune encephalomyelitis. Int Immunol 2016; 28(9):463-9.
- Edwards SC, Higgins SC, Mills KH. Respiratory infection with a bacterial pathogen attenuates CNS autoimmunity through IL-10 induction. Brain Behav Immun. 2015; 50: 41-46.
- Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005; 169: 425-434.
- Martinez J, Almendinger J, Oberst A, Ness R, Dillon CP, Fitzgerald P. Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc Natl Acad Sci U S A. 2011; 108: 17396-17401.
- Kanayama M, He YW, Shinohara ML. The lung is protected from spontaneous inflammation by autophagy in myeloid cells. J Immunol. 2015; 194: 5465-5471.
- Abdel Fattah E, Bhattacharya A, Herron A, Safdar Z, Eissa NT. Critical role for IL-18 in spontaneous lung inflammation caused by autophagy deficiency. J Immunol. 2015; 194: 5407-5416.
- Hirota K, Yoshitomi H, Hashimoto M, Maeda S, Teradaira S, Sugimoto N. Preferential recruitment of CCR6-expressing Th17 cells to inflamed joints via CCL20 in rheumatoid arthritis and its animal model. J Exp Med. 2007; 204: 2803-2812.
- Arima Y, Harada M, Kamimura D, Park JH, Kawano F, Yull FE. Regional neural activation defines a gateway for autoreactive T cells to cross the blood-brain barrier. Cell. 2012; 148: 447-457.
- Reboldi A, Coisne C, Baumjohann D, Benvenuto F, Bottinelli D, Lira S. C-C chemokine receptor 6-regulated entry of TH-17 cells into the CNS through the choroid plexus is required for the initiation of EAE. Nat Immunol. 2009; 10: 514-523.
- Yamazaki T, Yang XO, Chung Y, Fukunaga A, Nurieva R, Pappu B. CCR6 regulates the migration of inflammatory and regulatory T cells. J Immunol. 2008; 181: 8391-8401.
- Pittas F, Ponsonby AL, van der Mei IA, Taylor BV, Blizzard L, Groom P, et al. Smoking is associated with progressive disease course and increased progression in clinical disability in a prospective cohort of people with multiple sclerosis. J Neurol. 2009; 256: 577-585.
- Handel AE, Williamson AJ, Disanto G, Dobson R, Giovannoni G, Ramagopalan SV. Smoking and multiple sclerosis: an updated meta-analysis. PLoS One. 2011; 6: e16149.
- Meuronen A, Majuri ML, Alenius H, Mäntylä T, Wolff H, Piirilä P, et al. Decreased cytokine and chemokine mRNA expression in bronchoalveolar lavage in asymptomatic smoking subjects. Respiration. 2008; 75: 450-458.
- Panitch HS. Influence of infection on exacerbations of multiple sclerosis. Ann Neurol. 1994; 36 Suppl: S25-28.
- Edwards S, Zvartau M, Clarke H, Irving W, Blumhardt LD. Clinical relapses and disease activity on magnetic resonance imaging associated with viral upper respiratory tract infections in multiple sclerosis. J Neurol Neurosurg Psychiatry. 1998; 64: 736-741.
- Kriesel JD, White A, Hayden FG, Spruance SL, Petajan J. Multiple sclerosis attacks are associated with picornavirus infections. Mult Scler. 2004; 10: 145-148.