
Maternally Inherited Huntington Disease and Paternally Inherited Neurofibromatosis Type-1 In an Individual- A Rare Co-Occurrence
Ashutosh Gupta1*, Suman Kushwaha2
1Department of Neurology, Institute of Human Behavior and Allied Sciences, Delhi, India
Introduction
Huntington disease (HD) is a neuro degenerative disorder caused by an autosomal dominantly inherited CAG trinucleotide expansion in huntingtin (HTT) gene on chromosome 4 p16.3, affecting both men and women equally. Mutation in HTT gene results in abnormally long polyglutamine repeats. The sex of the transmitting parent is the major determinant of intergenerational changes. In paternal transmission, the CAG repeats often increases from generation to generation (anticipation phenomenon). Whereas affected mothers almost never transmit a significantly expanded CAG repeats. Contrarily, contractions do occur more frequently in the offsprings1. HD is characterized by motor, behavioral, oculomotor and cognitive dysfunction. Typical onset of clinical symptoms being apparent in midlife (4th decade) but younger and older onsets also occur. As the disease progresses, chorea becomes more prominent leading to complete disability. Ultimately, death occurs within 15 - 20 years due to number of mechanisms. The prevalence of this disease is 10.6 to 13.7 individuals per 100 000 in western population and 1-7 individuals per million in Japan, Taiwan and Hongkong2. There is a dearth of data on HD prevalence in India. However, among the Indian population, the overall prevalence of HD is lower than that of Western countries3. The repeat CAG length of 26 and smaller is normal, 27-35 is not associated with disease expression, 36-39 is associated with reduced penetrance and 40 or larger is associated with disease expression.
Neurofibromatosis type 1 (NF-1) is caused by an autosomal dominant mutation in the NF-1 gene, located on chromosome 17q11.2. The prevalence is approximately 1 in 3000-4000 individuals. The occurrences are present in all the racial groups and affect both the sexes equally. The disease is characterized clinically by the presence of café au lait macules, neurofibromas, freckles, and Lisch nodules4. Neurofibromas can occur anywhere in the peripheral nervous system. In patients with NF-1, gliomas occur frequently in all parts of the brain, brain stem and cerebellum. Lisch nodules, which are innocuous iris hamartomas present in > 90% of adults with NF-15. NF-1 gene encodes for neurofibromin protein. The latter widely expresses in nervous system and reduces cell proliferation by accelerating the inactivation of a cellular proto-oncogene, p-21 Ras, which promotes fibroma and tumor formation6. Nearly 50% of children born with NF-1 disorder have speech problems, learning disabilities, seizures and hyperactivity.
Case Report
A 32-year-old female presented to the Movement Disorder Clinic, Institute of Human Behavior and Allied Sciences, New Delhi, India, with complaints of insidious onset of abnormal movements involving all the limbs and trunk for the last five years. These movements were involuntary, continuous, irregular, non-jerky and semi-purposeful (video 1). These movements were aggravated during stress and subsided during sleep. Initially, the movements were mild and noticed by the observers but gradually progressed such that the patient started to notice them. In the past year, she developed difficulty performing routine housework because of the movements and increasing disability. She also developed behavioral abnormalities for the past two years in the form of frequent outbursts of anger, disturbances in mood, usually depression that increased in frequency in the last six months.
http://jneurology.com/articles/videos/jnnm-19-1244.mov
Video 1: showing the patient with choreiform movements involving her trunk and limbs.
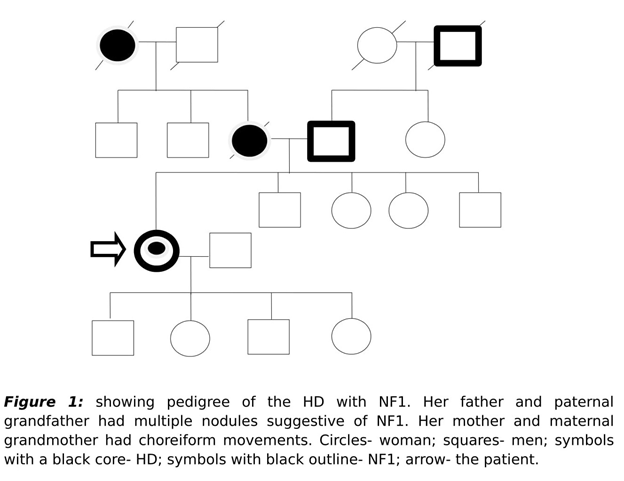
Family history was notable for her father and paternal grandfather had similar nodular lesions of varying sizes over their trunk and limbs. Her mother and maternal grandmother had similar kind of involuntary movements. Her mother also had behavioral abnormalities in the form of anger outbursts and suicidal ideations. All her siblings and children (aged 1,4,5 and 7 years) were asymptomatic. Out of 5 of her affected family members within 3 generations, 4 were afflicted either with HD or NF-1 and the patient had both HD and NF-1 (Figure 1).
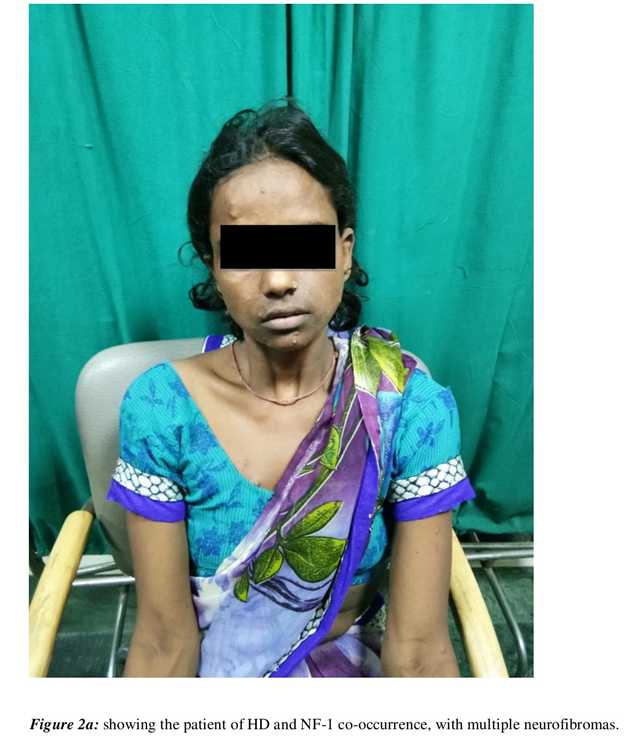
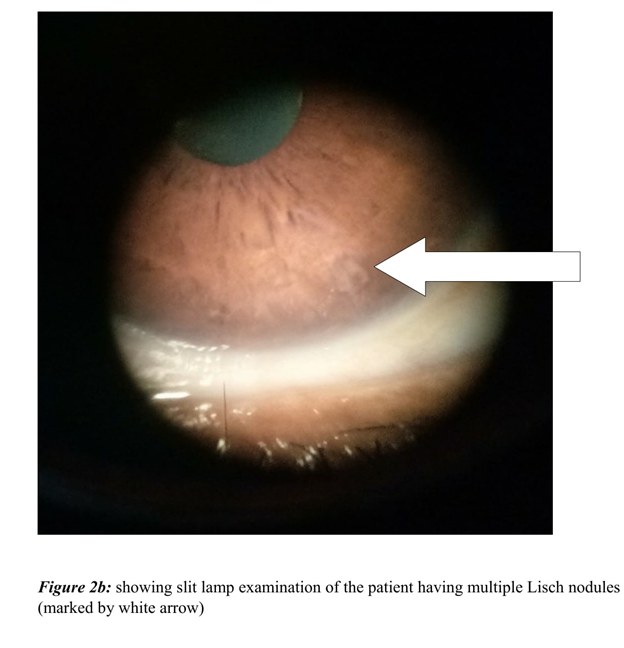
On examination, she had multiple non-tender nodules of varying sizes ranging from 5mm to 3 centimeters, distributed over face, limbs and trunk (Figure 2a). She reported that they were present since her childhood. Her slit lamp examination showed multiple Lisch nodules with no evidence of Kayer-Fleischer rings (Figure 2b). On CNS evaluation, she was alert and oriented and MMSE was 26. Her cranial nerves examination revealed prolonged saccadic latency in ocular examination. Also, no facial asymmetry, swallowing difficulty or dysarthria were discernable. She had reduced tone with apparently normal power in all the four limbs. There was generalized chorea with signs of motor impersistence. Her routine hematological, biochemical tests, radiological and neuro-imaging were non-revealing. Her DNA analysis revealed 48 CAG repeats, based on fragment analysis on ABI3730 XL and in cross reference with sequencing-based CAG length estimation on an optimized set of reference samples. Rest of her family members refused to undergo genetic test.
She was diagnosed to have both HD and NF-1 disorders. HD was confirmed by signs, symptoms and CAG repeat length of 48 while NF-1 was confirmed in accordance with the criteria of NIH. She was started on haloperidol at 1mg/d which was gradually increased to 5mg/d in follow up. There was a partial response for two months, but she continued to have subtle movements which lasted, thereafter.
Discussion
The present case is unique in two ways. Firstly, there was a linear inheritance of HD from maternal linage and secondly, there was a co-occurrence of both HD and NF-1 in an individual. To our knowledge this is the fourth case of co-occurrence in the world and first in Indian subcontinent.
In the present case, the multigenerational transmission of mutated HTT gene (mHTT), was inherited maternally. In maternal inheritance, the extrachromosomal organelles or cytoplasmic factors are exclusively transmitted unchanged from mother to off springs which may modify the onset or expression of mHTT gene. In our patient, the HD occurred as an adult onset resulting into generalized chorea with signs of motor impersistence. The off springs of affected mothers are likely to have either no change in CAG repeats or reduction in size of the CAG repeats1. Thereby, the other asymptomatic members of the family might have the size of CAG repeats in the non-expressive zone (27-35 CAG repeats). Moreover, many autosomal dominant gene mutations show varying degree of penetrance, most likely, due to influence of other genes or environmental factors4.
NF-1 is characterized by extreme clinical variability, not only among unrelated individuals but also among the affected individuals within a single family and even within a single person at different times in life5.
However, as found in our case, many patients will not have substantial problems other than the cutaneous fibromas including Lisch nodules and mild cognitive impairments6. NF-1 is caused by heterozygous mutations in NF-1 gene. Resultantly, 50% of the patients with NF-1, inherit the mutation from their affected parent while another 50% develop de novo. The NF-1 gene, being larger in size (>350 Kbp), is unstable and prone to higher spontaneous mutations, which may account for 50% de novo mutations6. In our case, there appears to be a whole NF-1 gene deletion, promoting large number of cutaneous neurofibromas. The mosaicism (individuals having two or more genetically different cell populations) is known to be caused by post zygotic NF-1 gene mutation4. Segmental mosaicisms are restricted to one part of the body only and are present in the individuals, whose parents are unaffected5. In mosaic or segmental NF-1 the constitutional genetic defect is present in only a small population of cells6.
In the present case, the patient inherited both HD and NF-1 disorders from her maternal and paternal lines, respectively. None of her parents and grandparents were afflicted with both HD and NF-1. The causative genes are located on two different chromosomes and by chance co-segregated in this individual. The NF-1 was apparent from her childhood while HD occurred at the age of 27 years as an adult onset and by 32 years of age the CAG repeats were 48. Only 3 studies have, so far, been published on the association of HD and NF-1 disorders7-9. Two of them reported autopsy findings of 58 and 60 years-old women and found only HD pathology8,9. However, the latter study found polyglutamine inclusions in all the layers of cerebral cortex and in amygdala, hippocampus and striatum, apart from presence of neuronal intranuclear inclusions in the cerebellum, which is not associated with the HD9. Sevick et al reviewed 68 MR images and found 49% of white matter lesions due to NF-1 in the cerebellum10. Further, the gliomas, caused by NF-1, are also reported to occur in all parts of the brain, brain stem and the cerebellum6. Thus, the pathological findings of NF-1 appear to be present in the cerebellum, in the concurrent cases of HD and NF-1 disorders, which is a matter of further investigation.
The association of NF-1 and HD disorders in a single individual is not fortuitous but suggesting that these two distinct disorders are in a linkage disequilibrium. So, there is likely to be presence of common pathogenic mechanism that may be due to the threshold effect of expanded polyglutamine. Remarkably, the present study as well as two of the three earlier studies8,9, found co-occurrence of HD and NF-1 in females only.
Conclusion
Although, both these genetic disorders are inherited via different mutations on different chromosomes, further investigations are needed in future to find out any confounding linkage between the pathophysiology of these two distinct diseases, particularly in females.
Acknowledgement
I would like to acknowledge Mr. S. M. Gupta, my father, and mentor for providing useful directions in making this report.
Ethics Statement and Statement of Informed Consent
Written permission was obtained from the patient in compliance with any laws regarding patient authorizations relating to the use or disclosure of protected health information of the jurisdiction to which the patient and the physician are subjected to. No experimental investigation of human subjects was done in this study.
References
- Kremer B, Almqvist E, Theilmann J, et al. Sex-Dependent Mechanisms For Expansions and Contractions of the CAG Repeat on Affected Huntington Disease Chromosomes. Am J Hum Genet. 1995; 57: 343-350.
- McColgana P, Tabrizi SJ. Huntington’s disease: a clinical review. European Journal of Neurology. 2018; 25: 24–34. doi:10.1111/ene.13413
- Pramanik S, Basu P, Gangopadhaya PK, et al. Analysis of CAG and CCG repeats in Huntingtin gene among HD patients and normal populations of India. European Journal of Human Genetics. 2000; 8(9): 678–682. doi:10.1038/sj.ejhg.5200515
- Kehrer-Sawatzki H, Cooper DN. Mosaicissm in sporadic neurofibromatosis type 1: variations on a theme common to other hereditary cancer syndromes. J Med Genet. 2008; 45: 622–631. doi:10.1136/jmg.2008.059329
- Kimberly Jett, Jan M. Friedman. Clinical and genetic aspects of neurofibromatosis 1. Genetics IN Medicine • Volume 12, Number 1, January 2010. DOI: 10.1097/GIM.0b013e3181bf15e3
- Rosalie E Ferner. Neurofibromatosis 1 and neurofibromatosis 2: a twenty first century perspective. Lancet Neurol. 2007; 6: 340–51.
- Pearson JS, Petersen MC. Coincidence of Huntington’s Chorea and multiple neurofibromatosis in two generations. Am J Hum Genet. 1954; 6: 344–345.
- Dörstelmann D, Kerschensteiner M, Markus E, et al. Huntington’s chorea and neurofibromatosis (von Recklinghausen’s disease). Concurrence of two generations of a family with inherited Huntington’s chorea. Z Neurol. 1971; 199: 39–45.
- Ito KAWAKAMI, Omi KATSUSE, Naoya AOKI, et al. Autopsy case of concurrent Huntington’s disease and neurofibromatosis type 1. PSYCHOGERIATRICS. 2014; 14: 81–86. DOI:10.1111/psyg.12040.
- Robert J, Sevick A, James Barkovich MSB, et al. Evolution of white matter lesions in neurofibromatosis 1: MR Findings. AJR. July 1992; 159: 171-175.