
MCPIP mediates preconditioning protection against ischemia
Jianli Niu and Pappachan E. Kolattukudy*
Abstract
Inflammatory response represents one of the first immune processes following injury. However, evidence indicates that inflammatory response can also induce cellular protection associated with preconditioning, a phenomenon in which brief episodes of a sublethal insult induce robust protection against subsequent lethal injuries. The elucidation of mechanisms that allow inflammatory response to confer cellular protection is critical to developing new therapeutic strategies against acute ischemic insults. In the present review, we will give a short overview on a novel zinc-finger protein, MCPIP (also known as Zc3h12a or Regnase-1), which may function as a master integrator of endogenous cellular protection exerted by preconditioning.
Introduction
Exposure to inflammatory agents is known to protect organs such as heart and brain against subsequent ischemic insults. This protection is called preconditioning, a phenomenon which brief episodes of a sublethal insult induce robust protection against subsequent lethal injuries. For example, cardiomyocyte-targeted expression of monocyte chemotactic protein-1 (MCP-1) showed such a preconditioning protective effect against experimental myocardial ischemic injury1,2. Coronary occlusion followed by reperfusion showed that the animals expressing MCP-1 had a much smaller infarct size when compared to the wild type1. Evidence suggested that SAPK/JNK activation was involved in the preconditioning effects1. Prevention of left ventricle (LV) dysfunction and remodeling in the mice with cardiomyocyte-targeted expression of MCP-1 was found to involve enhanced neovascularization and promotion of differentiation of cardiac fibroblasts into myofibroblasts2. Brain preconditioning by inflammatory inducer has also been reported3-5. In a mouse model of middle cerebral artery occlusion (MCAO) preconditioning by low dose of LPS attenuated infarct volume6. Preconditioning can be caused by a variety of inflammation-inducing treatments including prior ischemia, treatment with LPS or other Toll-like receptor (TLR) ligands3-5. Thus, the elucidation of mechanisms that allow inflammatory response to confer cellular protection is critical to developing new therapeutic strategies against acute ischemic insults.
MCPIP in Preconditioning of the Brain
The preconditioning protective effect afforded by prior exposure to the inflammatory agents suggested that the inflammatory agents induced the formation of a protective agent6-11. Evidence was presented that MCPIP, a zinc-finger protein, first identified as a novel protein induced by MCP-1 treatment of human peripheral blood monocytes, is involved in the preconditioning6. Preconditioning of brain against ischemia damage by treatment of mice with low levels of LPS was found to be mediated via MCPIP. Thus, the brain of mice treated with low levels of LPS showed preconditioning protective effect against ischemia caused by transient MCAO. This protection was accompanied by induction of MCPIP. MCPIP transcript and protein levels were found to be enhanced by treatment of mouse and human microglia with low levels of LPS6. MCPIP deficient mice showed significant increase in infarct volume caused by MCAO and deficiency in LPS preconditioning. Mortality caused by LPS treatment of mice was significantly higher in MCPIP deficient mice. Probably the increased inflammation manifested in MCPIP deficient mice involves activation of C-Jun N-terminal kinase (JNK) as inactivation of JNK signaling decreased the production of inflammatory cytokines in MCPIP deficient mice after MCAO6.
MCPIP in Preconditioning of the Heart
LPS induced preconditioning in the heart was also found to be mediated via MCPIP. The level of MCPIP was significantly increased in the murine heart after LPS challenge12. Cardiomyocyte-targeted expression of MCPIP protected against endotoxin-induced myocardial inflammation and dysfunction12. The level of MCPIP was also significantly increased in the murine heart after ligation of left coronary artery13. Mice with cardiomyocyte-targeted expression of MCPIP showed improved survival, decreased cardiac hypertrophy, lower fibrosis and scar formation as well as better left ventricular function after myocardial infarction13. MCPIP overexpressing murine heart also showed decreased monocytic cell infiltration and inflammatory cytokine expression as well as lower caspase 3/7 activities and apoptotic cell death compared to wild type mice13.These protective effects of MCPIP probably involve its ability to inhibit NF-kB activation as the MCPIP expressing murine hearts showed lower NF-kB signaling12,13. The anti-Dicer activity of MCPIP is also involved in its protection against ischemic damage as the MCPIP expressing murine hearts showed lower expression of inflammation-associated miR-126,-1461,-155 and -199a when compared to the post-infarct hearts of the wild type mice13.
MCPIP in Pharmacological Preconditioning
Minocycline, a tetracycline antibiotic, shows anti-inflammatory neuroprotective effects independent of its anti-microbial activity14. It offers advantages as a candidate for treatment for stroke because of its superior penetration of the brain tissue prolonged treatment window and good chemical safety14,15. The neuroprotective effect of minocycline against ischemic stroke is mediated via MCPIP16. Thus, minocycline treatment of mice induced MCPIP in the brain and attenuated MCAO-induced infarct volume, neurological deficits and enhancement of inflammatory cytokine production. Such a monocycine-dependent protection was not seen in MCPIP-deficient mice. Minocycline induced MCPIP production was also seen in vitro in primary neuron-glial cells and cortical neurons16. Minocycline-induced protection was not found in MCPIP deficient neuron-glial cells and cortical neurons against reduced oxygen glucose deprivation induced cell death16. It is likely that the widely recognized anti-inflammatory property of minocycline is mediated via MCPIP.
MCPIP in Electroacupuncture Preconditioning
Tolerance to focal ischemia is also induced by pretreatment with electroacupuncture (EA)17. Ischemia tolerance consists of an early phase that occurs within minutes after ischemia induction followed by a delayed phase that lasts for hours or even days. Mechanisms underlying delayed preconditioning that produces a potent neuroprotection, may suggest novel treatment for ischemic stroke18,19. EA pretreatment-induced neuroprotection is related to the suppression of inflammatory processes in the ischemic area, and this suppression is probably mediated via induction of MCPIP19. Thus, EA pretreatment at the baihui acupoint induced MCPIP production and attenuated MCAO-induced inflammatory response and ischemic injury19. Pretreatment with EA attenuated infarct volume, leukocyte infiltration into the brain, production of inflammatory cytokines and neurological deficits. MCPIP deficient mice did not demonstrate the EA pretreatment-induced protection. NF-kB activation in the brain resulting from MCAO was attenuated by EA pretreatment, but this attenuation was not found in MCPIP deficient mice19. Thus, MCPIP is involved in mediating the EA pretreatment –induced delayed brain ischemia tolerance. Since pretreatment is not a practical method for treatment of stroke it was tested whether EA treatment after stroke could offer therapeutic benefits. In fact treatment with EA after MCAO afforded protection as indicated by attenuated infarct volume19.
Molecular Mechanisms of MCPIP-Mediated Preconditioning
The molecular mechanisms underlying the protection against ischemic stroke afforded via MCPIP remain to be elucidated. The potent anti-inflammatory function of MCPIP is mediated in part by its inhibition of NF-kB activation via deubiquination of TRAF proteins and other polyubiquinated components required for activation of the kinase that phosphorylates IkB to cause its ubiquitination and degradation by the proteasome system20. Degradation of IkB releases the active subunits of NF-kB that enters the nucleus and transcriptionally activate the genes encoding inflammatory molecules such as cytokines. In addition to this down regulation of inflammation via the deubiquinase activity of MCPIP, the RNase activity of MCPIP destabilizes and causes degradation of the transcripts of inflammatory cytokines such as IL-1k and IL-6 by binding to the stem loop structure at 3’-UTR of the mRNA21. MCPIP can also degrade 3’-UTR-independent degradation of mRNA of the receptors of inflammatory cytokines such as IL-1722. Even though RNase activity–dependent degradation of IL-17 receptor transcript was shown to be independent of its UTR, the structural features of the transcript involved in the binding and degradation of IL-17 receptors remain unknown. Damage-associated molecule patterns (DAMPs), known also as alarmins, signal tissue damage. High motility group box 1 (HMGB1), normally located in the nucleus, is secreted into the extracellular medium upon stroke injury23. HMGB1 binds to its receptor and triggers potent inflammatory response via activation of NF-kB23. HMGB1 induces MCPIP in microglia cell line and in microglia in rat brain24. Knockdown of MCPIP exacerbated inflammation and neuronal injury suggesting that MCPIP plays a central role in limiting HMGB1-induced inflammation24. The role of MCPIP in the protection against stroke damage was demonstrated with mice with neuronal targeted deletion and mice with neuron –targeted overexpression of MCPIP. The neuron-specific absence of MCPIP caused a larger infarct volume and enhanced production of inflammatory cytokines when compared to wild type animals subjected to MCAO. On the other hand, neuron-specific overexpression of MCPIP attenuated ischemic damage caused by MCAO manifesting smaller infarct volume and suppressed production of inflammatory cytokines when compared to wild type mice (J. Liang and P.E. Kolattukudy, unpublished).
Prior treatment with inflammation inducing conditions such as early periods of ischemia and LPS protect against subsequent ischemic insults in tissues such as brain and heart. All of these preconditioning effects are probably mediated via induction of MCPIP. The preconditioning protective effect of minocycline treatment is also mediated via induction of MCPIP. MCPIP mediates the preconditioning protective effects most probably using both its RNase activity and deubiquitinase activity that make MCPIP a potent anti-inflammatory agent (Figure 1). Deubiquiniase activity inhibits NF-kB activation and probably regulates the ubiquitination state of proteins that can determine their subcellular localization, function and fate. The RNase activity destabilizes and enhances degradation of mRNA for inflammatory cytokine and their receptors21,22. The anti-Dicer RNase activity inhibits synthesis of microRNAs (miRs) that can influence cellular function in many ways25. Each of the nearly 2000 miRs that are expressed in a tissue-specific manner can target multiple mRNA and each mRNA can be targeted by multiple miRs. miRs can regulate the formation of other miRs making a very complex picture of possibilities for regulation of preconditioning and other biological processes. Comprehensive studies on the effects of MCPIP on the regulation of ubiquitome and miRome will be required to reveal the ways in which MCPIP regulates cellular function.
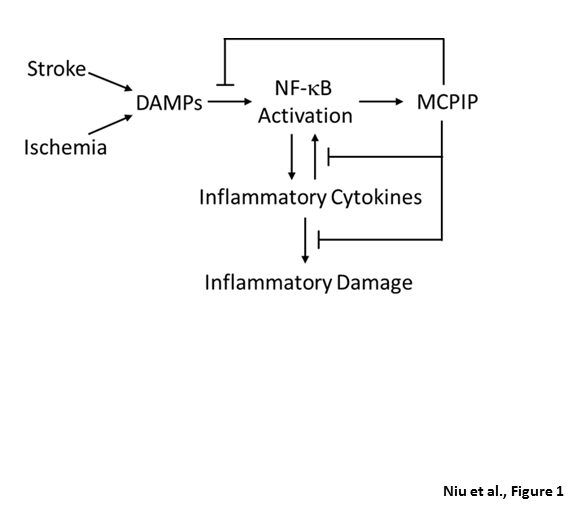
Figure 1: The role of MCPIP in preconditioning. Ischemia- and stroke-induced signals cause activation of NF-kB that leads to transcriptional activation of inflammatory cytokines that can in turn stimulate NF-kB activation causing induction of MCPIP that can inhibit NF-kB activation via deubiquiniase activity and its RNase activity enhances degradation of transcripts for inflammatory cytokines and their receptors, and inhibits miR synthesis.
Clinical Aspects
Stroke and cognitive deficits resulting probably from microembolism accompany coronary bypass grafting, albeit in a small subset of patients. Pharmacological preconditioning strategy is being assessed in several clinical trials4. Pre-infarction angina (spontaneous ischemia) prior to acute myocardial infarction makes the infarction milder with fewer cardiac arrhythimias and LV dysfunction26. Retrospective studies of stroke showed that previous transient ischemia attack attenuates the severity of subsequent stroke with better outcome compared to patients without previous events27,28. These demonstrations constitute indications that suggest preconditioning occurs in human brain although there are studies that failed to detect preconditioning benefits29. The clinical application of preconditioning still remains to be made practical. Even though MCPIP appears to be a key player in stroke injury as a potent neuroprotection agent, it is not known whether MCPIP can be used for treatment of stroke. It is possible that administration of MCPIP protein or MCPIP expressing DNA construct or virus directly to the infarct area soon after stroke may help to suppress inflammation and attenuate the ischemic damage. However, such a possibility has not been experimentally tested.
Conclusion
Accumulating evidence suggests that MCPIP is involved in a number of physiological processes as well as in the pathogenesis of neuron and cardiovascular diseases. The use of gene knockout and transgenic animals has made a significant contribution to understanding the different mechanisms regulated by MCPIP. Based on current knowledge of MCPIP, it is conceivable that MCPIP should be one of the prime targets in the discovery of drugs to combat neuron and cardiovascular disorders in which inflammation plays a significant role.
Acknowledgements
The study was supported in part by Grant HL-69458 from National Institutes of Health.
References
- Martire A, Fernandez B, Buehler A, Strohm C, Schaper J, Zimmermann R, et al. Cardiac overexpression of monocyte chemoattractant protein-1 in transgenic mice mimics ischemic preconditioning through SAPK/JNK1/2 activation. Cardiovasc Res. 2003;57:523-534.
- Morimoto H, Takahashi M, Izawa A, Ise H, Hongo M, Kolattukudy PE, et al. Cardiac overexpression of monocyte chemoattractant protein-1 in transgenic mice prevents cardiac dysfunction and remodeling after myocardial infarction. Circ Res. 2006;99:891-899.
- Rosenzweig HL, Lessov NS, Henshall DC, Minami M, Simon RP, Stenzel-Poore MP. Endotoxin preconditioning prevents cellular inflammatory response during ischemic neuroprotection in mice. Stroke. 2004;35:2576-2581.
- Dirnagl U, Becker K, Meisel A. Preconditioning and tolerance against cerebral ischaemia: from experimental strategies to clinical use. Lancet Neurol. 2009;8:398-412.
- Gesuete R, Kohama SG, Stenzel-Poore MP. Toll-like receptors and ischemic brain injury. J Neuropathol Exp Neurol. 2014;73:378-386.
- Liang J, Wang J, Saad Y, Warble L, Becerra E, Kolattukudy PE. Participation of MCP-induced protein 1 in lipopolysaccharide preconditioning-induced ischemic stroke tolerance by regulating the expression of proinflammatory cytokines. J Neuroinflammation. 2011;8:182.
- Stowe AM, Wacker BK, Cravens PD, Perfater JL, Li MK, Hu R, et al. CCL2 upregulation triggers hypoxic preconditioning-induced protection from stroke. J Neuroinflammation. 2012;9:33.
- Wacker BK, Perfater JL, Gidday JM. Hypoxic preconditioning induces stroke tolerance in mice via a cascading HIF, sphingosine kinase, and CCL2 signaling pathway. J Neurochem. 2012;123:954-962.
- Xu X, Kim JA, Zuo Z. Isoflurane preconditioning reduces mouse microglial activation and injury induced by lipopolysaccharide and interferon-gamma. Neuroscience. 2008;154:1002-1008.
- Marsh B, Stevens SL, Packard AE, Gopalan B, Hunter B, Leung PY, et al. Systemic lipopolysaccharide protects the brain from ischemic injury by reprogramming the response of the brain to stroke: a critical role for IRF3. J Neurosci. 2009;29:9839-9849.
- Kaucsár T, Bodor C, Godó M, Szalay C, Révész C, Németh Z, et al. LPS-induced delayed preconditioning is mediated by Hsp90 and involves the heat shock response in mouse kidney. PLoS One. 2014;9:e92004.
- Niu J, Wang K, Graham S, Azfer A, Kolattukudy PE. MCP-1-induced protein attenuates endotoxin-induced myocardial dysfunction by suppressing cardiac NF-?B activation via inhibition of I?B kinase activation. J Mol Cell Cardiol. 2011;51:177-186.
- Niu J, Jin Z, Kim H, Kolattukudy PE. MCP-1-induced protein attenuates post-infarct cardiac remodeling and dysfunction through mitigating NF-κB activation and suppressing inflammation-associated microRNA expression. Basic Res Cardiol. 2015;110:26./li>
- Stock ML, Fiedler KJ, Acharya S, Lange JK, Mlynarczyk GS, Anderson SJ, et al. Antibiotics acting as neuroprotectants via mechanisms independent of their anti-infective activities. Neuropharmacology. 2013;73:174-182.
- Garrido-Mesa N, Zarzuelo A, Gálvez J. Minocycline: far beyond an antibiotic. Br J Pharmacol. 2013;169:337-352.
- Jin Z, Liang J, Wang J, Kolattukudy PE. MCP-induced protein 1 mediates the minocycline-induced neuroprotection against cerebral ischemia/reperfusion injury in vitro and in vivo. J Neuroinflammation. 2015;12:39.
- Wang Q, Peng Y, Chen S, Gou X, Hu B, Du J, et al. Pretreatment with electroacupuncture induces rapid tolerance to focal cerebral ischemia through regulation of endocannabinoid system. Stroke. 2009;40:2157-2164.
- Zhao H, Ren C, Chen X, Shen J. From rapid to delayed and remote postconditioning: the evolving concept of ischemic postconditioning in brain ischemia. Curr Drug Targets. 2012;13:173-187.
- Jin Z, Liang J, Wang J, Kolattukudy PE. Delayed brain ischemia tolerance induced by electroacupuncture pretreatment is mediated via MCP-induced protein 1. J Neuroinflammation. 2013;10:63.
- Liang J, Saad Y, Lei T, Wang J, Qi D, Yang Q, et al. MCP-induced protein 1 deubiquitinates TRAF proteins and negatively regulates JNK and NF-kappaB signaling. J Exp Med. 2010;207:2959-2973.
- Matsushita K, Takeuchi O, Standley DM, Kumagai Y, Kawagoe T, Miyake T, et al. Zc3h12a is an RNase essential for controlling immune responses by regulating mRNA decay. Nature. 2009;458:1185-1190.
- Garg AV, Amatya N, Chen K, Cruz JA, Grover P, Whibley N, et al. MCPIP1 Endoribonuclease Activity Negatively Regulates Interleukin-17-Mediated Signaling and Inflammation. Immunity. 2015;43:475-487.
- Singh V, Roth S, Veltkamp R, Liesz A. HMGB1 as a Key Mediator of Immune Mechanisms in Ischemic Stroke. Antioxid Redox Signal. 2016;24:635-651.
- Liu XX, Wang C, Huang SF, Chen Q, Hu YF, Zhou L, et al. Regnase-1 in microglia negatively regulates high mobility group box 1-mediated inflammation and neuronal injury. Sci Rep. 2016;6:24073.
- Suzuki HI, Arase M, Matsuyama H, Choi YL, Ueno T, Mano H, et al. MCPIP1 ribonuclease antagonizes dicer and terminates microRNA biogenesis through precursor microRNA degradation. Mol Cell. 2011;44:424-436.
- Hashimoto A, Nakata T, Wakabayashi T, Yuda S, Eguchi M, Sasao H, et al. Limitations of spontaneous reperfusion and conventional medical therapy to afford myocardial protection through antecedent angina pectoris in acute myocardial infarction. Ann Nucl Med. 1999;13:337-346.
- Wegener S, Gottschalk B, Jovanovic V, Knab R, Fiebach JB, Schellinger PD, et al. MRI in Acute Stroke Study Group of the German Competence Network Stroke. Transient ischemic attacks before ischemic stroke: preconditioning the human brain? A multicenter magnetic resonance imaging study. Stroke. 2004;35:616-621.
- Moncayo J, de Freitas GR, Bogousslavsky J, Altieri M, van Melle G. Do transient ischemic attacks have a neuroprotective effect? Neurology. 2000;54:2089-2094.
- Dezfulian C, Garrett M, Gonzalez NR. Clinical application of preconditioning and postconditioning to achieve neuroprotection. Transl Stroke Res. 2013;4:19-24.