
Parkinsonism in both members of married couples
A.H. Rajput*, MBBS, FRCPC; A. Rajput, MD, FRCPC
Saskatchewan Movement Disorders Program, University of Saskatchewan/ Saskatoon Health Region
Abstract
Parkinsonism in both spouses has been reported in only 20 couples in the literature so far. Six of the studies included only one or two couples, but one study reported nine couples. Fifteen of the couples reported by others consisted of only clinical data. By contrast, our study of five couples had detailed clinical, pathological and genetic observation on all ten individuals. We found no evidence of person-to-person transmission of parkinsonism. Details of that study are provided in this review.
The literature evidence to date indicates that neither Parkinson’s disease nor other common parkinson variants – multiple system atrophy or progressive supranuclear palsy are transmitted by sexual or close personal contact in the married couples. As well, these syndromes are not based on shared environments or same genetic mutation.
The best explanation for parkinsonism in both partners of non-consanguineous couples is, that Parkinson syndrome in each spouse is a coincidental disorder
Background (Introduction)
Parkinson syndrome (PS) also known as parkinsonism is a clinical syndrome. It is characterized by the presence of at least two of the three symptoms – bradykinesia, rigidity and tremor1-4. Several degenerative disorders are associated with PS5, the most common being Parkinson’s disease (PD). PD is characterized by marked substantia nigra (SN) neuronal loss and Lewy body (LB) inclusion1,5,6. The next most common degenerative variants of PS are multiple system atrophy (MSA) and progressive supranuclear palsy (PSP)5,7. Definite diagnosis of the degenerative PS variants is based on pathological findings1,8-10. Several different gene mutations may manifest LB pathology similar to the PD11. Therefore, genetic testing adds to the understanding of PS pathophysiology11,12.
Well-known secondary causes of PS includes encephalitis lethargica (1915-1930)13, known as post-encephalitic parkinsonism (PEP)13-15. There have been no new cases of PEP after 195414.Methyl-phenyl-tetrahydropyridine (MPTP), a synthetic narcotic, leads to SN damage resulting in parkinsonism16. Rotenone, another tonic, is known to produce parkinsonian pathology17. Neuroleptics and some other medications can produce drug induced parkinsonism (DIP). The DIP cases do not have any histological abnormality in the brain18.
Possibility of person-to-person transmission
Based on the spread of host PD pathology into the transplanted normal fetal nigral tissue, it is conceivable that PD may be transmitted from person-to-person19-22. However, there is no literature indicating such transmission from person-to-person. The mechanism of spread of the host PD pathology to normal fetal tissue transplant is not known23. It has been postulated that PD has some characteristics of prion disease22-24, and hence may be transmissible from person-to-person.
Expected pathology in transmitted PS
Both PD and MSA have alpha-synuclein positive inclusions – neuronal inclusions in PD and glial inclusions in MSA. In vivo studies indicate that transmission of each of those produces pathology similar to that of the donor. Animals transplanted PD alpha-synuclein develop PD pathology, while MSA transplants develop MSA-like pathology25. Hence, if either of those were transmitted from one person to another individual, the pathology in the host would be the same as in the donor.
Tau positive inclusions are seen in Alzheimer’s disease (AD), corticobasal degeneration (CBD) and the PSP, but they all are different strains of tau protein. Each of those tau strains when transplanted in animals produce changes identical to the tau pathology in the donor26. Transmission of a tauopathy in human subjects is therefore expected to produce the same type of tauopathy in the recipient as in the donor.
Married or otherwise cohabitating couples that have the most intimate personal contact offer unique opportunity to study person-to-person transmission of PS.
Optimal study to determine person-to-person transmission
Errors in clinical diagnosis of PS variants are well known1,9,10. Functional imaging studies to determine the integrity of the SN are valuable but they cannot distinguish between PD and other PS variants with more widespread pathology27,28. Additionally, the findings of functional imaging studies have not yet been confirmed with pathological evidence10,29. Neuropathology remains the gold standard for diagnosis of degenerative variants of PS30. Recent advances in genetics show that several genetic mutations can produce PD like pathology11. Therefore detailed clinical, pathological and genetic studies of each spouse are needed to determine person-to-person transmission.
Literature on conjugal PS
There is very limited literature on conjugal PS and most of it consists of clinical case reports of one or two couples31-36. Strang32reported two conjugal PS couples from Sweden. The wife in the first couple had a strong family history of Parkinson’s disease but the husband had sporadic PD. Both spouses in the second couple had history of encephalitis and a diagnosis of PEP. Thus, these four cases had three different causes – genetic, sporadic, and post-encephalitic PS32. The author concluded that the PS in these couples was “purely by chance”. Counihan34 reported one PS couple but provided no details. Miwa and Kondo33 reported one PS couple; the wife was clinically diagnosed as PD, and the husband had a clinical diagnosis of MSA. Ramani et al35 reported one PS couple. The husband had diagnosis of PD and the wife was clinically diagnosed as dementia with LB.
The largest series was reported by Willis et al31. This study included nine PS couples. The average married life before onset of PS was 40 years. All except one of the 18 cases in the study were clinically diagnosed as PD. They provided clinical details, occupation and analysis of common environmental exposure to pesticides and chemicals. Because the dates of onset in their cases31 were widely separated, they concluded that common environmental cause did not account for the PS in these couples.
None of the above noted 15 couples had pathological verification of the PS variants or genetic analysis31-35.
Rajput et al36 identified eleven conjugal PS couples from their clinic and published the most detailed clinical, pathological and genetic study in five married couples. They excluded two couples where only one spouse came to autopsy and four couples where neither spouse has come to autopsy. That study was restricted to five couples where both spouses came to autopsy and had genetic studies.
Saskatchewan Movement Disorders Program (SMDP) is a fully integrated specialized clinic and research set-up established in 1968. Every patient is seen at each visit by either or both movement disorders neurologists (AHR and AR)37. Special emphasis at the program is the longitudinal clinical follow-up and autopsy studies of those cases38. As a rule, the patients are seen at the MDCS at 6 to 12 month intervals. Videos are made on all consenting subjects. The patients seen at Movement Disorders Clinic Saskatchewan (MDCS) are offered a choice of autopsy study at no cost to family/estate. Autopsies are restricted to patients seen at MDCS37.
The final diagnosis is made by the treating neurologist based on all the available clinical and pathology information and where available genetic analysis. Details of the SMDP have been reported previously37,38.
Figure 1 shows the flow chart of the SMDP.
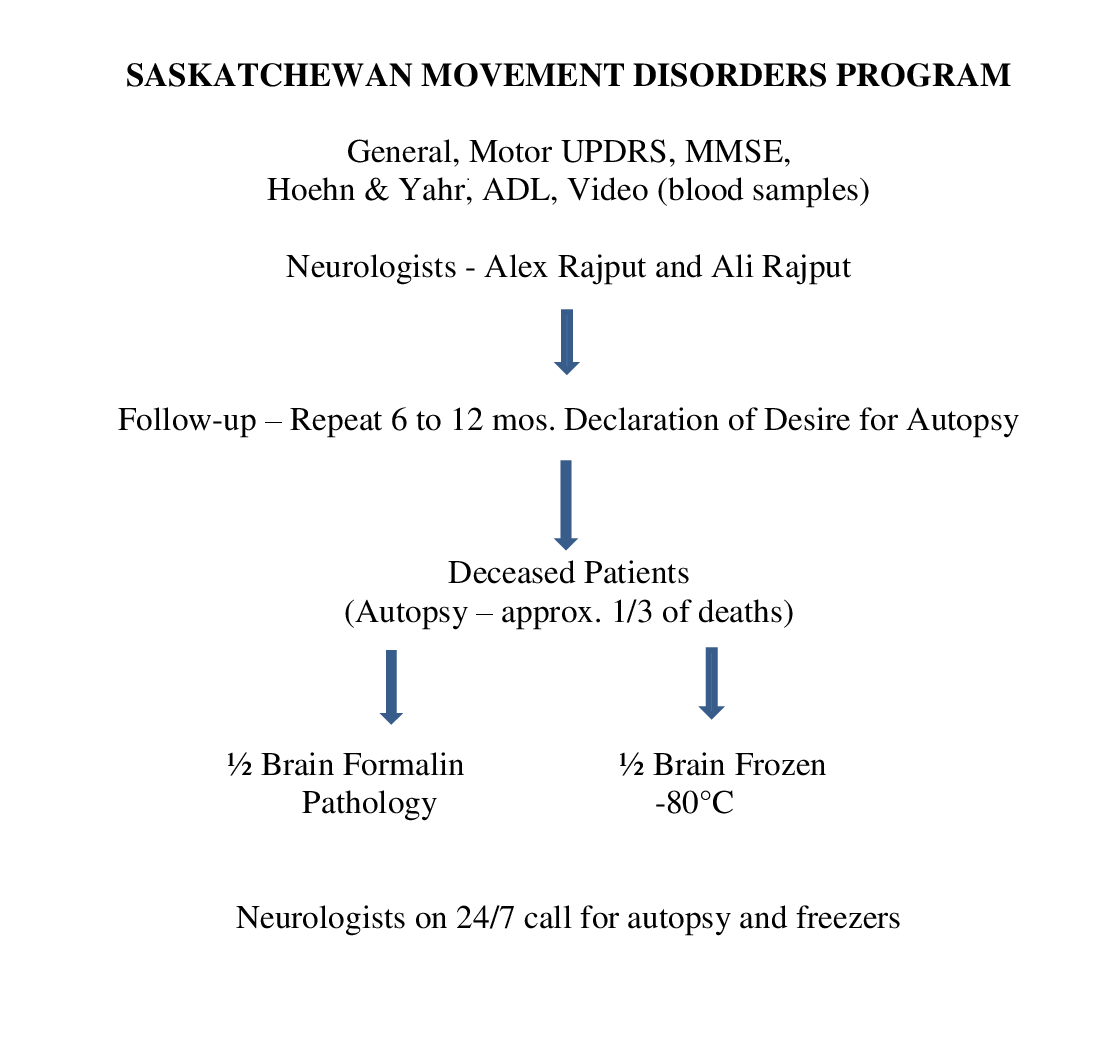
Figure 1:Flow chart of Saskatchewan Movement Disorders Program operations.
Clinical records, videos, half frozen brain, remnants for formalin fixed brain, paraffin blocks and pathology slides are preserved in special laboratories (Figure 2). This brain repository is different from “brain bank” as it includes only those patients that were looked after at the MDCS. 553 autopsies have been so far performed on the MDCS assessed cases - most had PS. This unique set-up permits studies of many important issues which cannot be adequately studied by other methods1,36,38-40. De-identified brain material is provided free of charge to research collaborators at major institutions around the world41-44.

Figure 2:Filing cabinet containing hard copy of patient clinical records. Picture 2. -80oC freezers. Currently there are nine freezers. Picture 3. Cardboard boxes, each containing half-frozen brain from a patient. Each box has patient identification at four places – two with only the number and two with name and number. Picture 4. Formalin-fixed remains of the brain tissue after pathology has been completed. Picture 5. Paraffin blocks and glass slides stored in our laboratory. Picture 6. Video library. (Reproduced from Rajput et al36 with permission from Cambridge University Press)
All 10 conjugal PS cases reported36 had video recordings made (see video). None of the couples were consanguineous or lived in the same close community during early age. Mean age of PS onset was 70.8 (60-80) years. Mean duration of marriage when the first spouse manifested PS was 44 (32-55) years. Details of cases are reported in Table 1.
| Gene | Relevant clinical information |
|---|---|
| Couple 1: Married 51 years at onset of PS in first spouse and 58 years at first spousal death. | |
|
F1 Housewife |
Onset at age 72 as balance difficulty with tendency to fall backwards. She was treated elsewhere with levodopa. First examined at age 80 when on levodopa. She reported subjective benefit. There was no WO, OO, or Dys. Her MMSE score was 30/30. She had square-wave jerks on forward gaze and slowed horizontal pursuit. Her motor profile was akinetic-rigid and overall disability was at Stage 4 H&Y. At age 82 she had dysphagia and needed care, had supranuclear ophthalmoplegia and she was at Stage 5 H&Y. Her MMSE was 30/30. Clinical diagnosis was PSP. Age at death was 85. |
|
M1 Farmer/Truck driver |
He had onset of right upper limb tremor at age 78. Examination at age 79 while on no drugs revealed mild parkinsonian features and was rated at Stage 2 H&Y. At age 84 he was started on levodopa which did not benefit and he discontinued on his own. At age 85 he was on no medication. He never had adequate trial on LD. He had no ophthalmoplegia or dystonia. The clinical diagnosis was PD. He died at age 85. (He had LRRK2 mutation. His 3 nieces also have LRRK2 mutation and PS. One of them came to autopsy with PD findings.) |
| Couple 2: Married 42 years at onset of PS in first spouse and 58 years at first spousal death. | |
|
F2 Housewife |
She had onset of gait difficulty at age 65. At age 70 she was receiving levodopa/carbidopa and selegiline. She had akinetic/rigid PS at Stage 3 H&Y. MMSE was 28/30. Over the course she was tried on LD, dopamine agonist, and amantadine without significant benefit. She had mild facial and upper limb dyskinesia on LD. Her extraocular movements remained normal. Final Clinical diagnosis was atypical PD. She died at age 84. |
|
M2 Police communication officer |
He had onset at age 68 with right upper limb tremor. Two years later he was at Stage 2 H&Y and his MMSE was 28/30. One year later he was at Stage 2.5 UPDRS. He had mixed motor clinical profile. He improved on LD. He never had WO, OO, or Dys. At age 79 he was at Stage 3. One year later he was in nursing home because of bilateral hip problem. Final clinical diagnosis was PD. He died at age 82. |
| Couple 3: Married 32 years at onset of PS in first spouse and 55 years at first spousal death. | |
|
F3 Housewife |
She had onset of right upper limb tremor at age 74. When evaluated at age 78 she had mixed motor profile and was at Stage 2.5 UPDRS. She was started on Sinemet and had mild dyskinesia and questionable WO but no OO. At age 78 her MMSE was 26/30. At age 80 she was at Stage 3 disability. The final clinical diagnosis was PD. She died at age 82. (Her one sister has pathology proven PD and another sister has clinical diagnosis of PD. Their son has clinical diagnosis of PD.) |
|
M3 Farmer |
He had onset of left upper limb functional decline at age 60. When examined at age 62 he was on no medication. He was at Stage 2 H&Y and had mixed motor profile. At age 65 he was at stage 3 H&Y and was started on LD with marked improvement. At age 70 he was at Stage 4 and at age 77 he was at stage 5. He had no Dys or OO but had mild WO. He had freezing of gait at age 79. There was no cognitive impairment until age 77. At age 79 he was in nursing home, had dementia and was at Stage 5 disability. His final clinical diagnosis was PD and dementia. He died at age 83. |
| Couple 4: Married 55 years at onset of PS in first spouse and 66 years at first spousal death. | |
|
F4 Teacher |
She had onset of generalized slowing at age 80. Examination at age 82 revealed akinetic/rigid PS at Stage 3 H&Y. She was receiving LD at that time. No reported WO, OO, or Dys. Her final clinical diagnosis was PD. She died at age 89. |
|
M4 Building maintenance worker |
He had onset of right upper limb tremor at age 77. When examined at age 81 he was receiving levodopa/carbidopa. There was no history of dyskinesia, WO or OO. He had mixed motor profile and MMSE was 30/30 and was rated at Stage 2.5 UPDRS. His final clinical diagnosis was PD. Age at death was 89. |
| Couple 5: Married 40 years at onset of PS in first spouse and 50 years at first spousal death. | |
|
F5 Housewife |
She had onset of decline in handwriting at age 66. On examination at age 67 she had akinetic/rigid PS and was rated at Stage 1.5 UPDRS. At age 68 she was at Stage 2, was having difficulty swallowing, had bladder urgency and her speech had significantly declined. Her MMSE was 30/30. At age 70 she was at Stage 4 H&Y. She was tried on LD but had no benefit. She had mild Dys but no WO or OO. At age 70 she had hallucinations on LD and the drug was discontinued. The final clinical diagnosis was MSA. She died at age 72. |
|
M5 Farmer |
He had onset of right upper limb tremor at age 68. Evaluation at age 70 he had mixed motor profile and was at Stage 2. At age 71 his MMSE was 30/30. He was treated with LD and had good response. He had mild WO but no Dys or OO. At age 71 his MMSE score was 29/30. At age 75 he was at Stage 3. His final clinical diagnosis was PD. He died at age 84. |
Table 1:WO=Wearing off; OO=On-off; Dys=Dyskinesia; PSP=Progressive Supranuclear Palsy; MSA=Multiple System Atrophy; CBD=Corticobasal degeneration; SN=Substantia nigra; PD=Parkinson’s disease; LD=levodopa; MMSE=Mini Mental State Examination; H&Y=Hoehn and Yahr Scale; UPDRS=Unified Parkinson’s Disease Rating Scale. (Reproduced from Rajput et al35 with approval from Elsevier).
Genetic studies were performed in the laboratory of Dr. M. Farrer at the University of British Columbia, Canada36. All subjects were screened for SNCA missense mutation and copy number variants, DNAJC13, LRRK2, SCA 2, 3, 12 and 17 nucleotides36,42. Only one individual – the male in couple #1 - had a genetic mutation. He had the LRRK2 p.G2019S mutation and has a strong family history of that mutation.
Final diagnosis and its significances to shared pathogenesis
In two couples (#3 and #4) each spouse had PD. The wife in couple #3 had strong family history of PD though genetic basis was not identified, but other three cases were sporadic. In couple #1, both spouses had tauopathy. However, the husband had a genetic basis - LRRK2 mutation tauopathy and a strong family history of the same mutation. The wife had clinical picture of PSP and neuropathology verification of that diagnosis. In couple #2, the wife had unclassified tauopathy but the husband had PD. In couple #5, the wife had MSA and the husband had PD. Table 2 shows the sequence of PS manifestation in these couples and the calendar year of onset in each case. The earliest PS onset was in 1976 and the most recent was in 2004. Although some cases had neurofibrillary tangle pathology none had a history or the clinical profile of PEP. There have been no new cases of PEP since 1954, and none of these cases36 fall in that category14.
| Couple # | Order of spouse to develop PS | Calendar year of onset | Pathology diagnosis |
|---|---|---|---|
| 1 |
1st M 2nd F |
1994 1995 |
Tauopathy LRRK2 positive (strong family history of same mutation) PSP |
| 2 |
1stF 2ndM |
1993 1995 |
Tauopathy (Unclassified) PD |
| 3 |
1stM 2ndF |
1976 1996 |
PD - DLB PD (strong family history of PD) |
| 4 |
1stM 2ndF |
2002 2004 |
PD PD |
| 5 |
1stM 2ndF |
1990 1993 |
PD MSA |
Table 2 – Sequence of PS onset in couples and PS subtype
1st = The spouse that manifested parkinsonism first; 2nd = Spouse that manifested parkinsonism subsequently M=Male; F=Female; PS=Parkinson syndrome; PSP=Progressive Supranuclear Palsy; PD=Parkinson’s Disease; MSA-Multiple System Atrophy; DLB=Dementia with Lewy body.
The final diagnosis of degenerative variant is based on pathological findings. Table 3 shows main distinguishing features of three major PS variants – PD, PSP, and MSA observed in these five couples.
| Parkinson Disease | Progressive Supranuclear Palsy | Multiple System Atrophy | ||
|---|---|---|---|---|
| 1 | Common mode of onset | Tremor (upper limb) | Motor slowing/Gait and balance difficulty | Bradykinesia/Gait difficulty |
| 2 | Symmetry of symptoms | Often asymmetrical | Often symmetrical | Often symmetrical |
| 3 | Rest tremor during course | Often present | Rare | Rare |
| 4 | Body posture | Flexed (Late) | Erect | Body & neck flexion (Early) |
| 5 | Autosomal dysfunction | Usually (Late) | Not prominent | Often early and pronounced |
| 6 | Corticospinal tract findings | Not a feature | Rare | More common |
| 7 | Cerebellar signs | Not a feature | Rare | More common |
| 8 | Supranuclear ophthalmoplegia | No | Yes (often) | No |
| 9 | Response to levodopa | Good | Modest in some cases | Modest in some cases |
| 10 | Survival | Longest | Intermediate | Shortest |
| 11 | Pathology |
Alpha-synuclein positive, Neuronal inclusions (Lewy body) |
Tau positive Neuronal inclusions |
Alpha-synuclein positive Glial inclusions |
Table 3 – Main characteristics of Parkinson disease, Progressive Supranuclear Palsy and Multiple System Atrophy
Based on the observation in these five couples, we will consider the etiological link of PS in the spouses.
Was PS transmitted from one partner to the other due to sexual or close social contact?
The transmission of PS from one partner to the other would produce the same pathology as in the donor25,26. It can be assumed that the spouse manifesting PS the earliest is the primary case that transmitted the disease to the other spouse. Table 2 shows that unclassified tauopathy of the wife in couple #2 did not produce the same pathology in the husband who had PD. In couple #5 the husband manifested PD the earliest, but the wife had MSA. In couple #1 the etiological basis of tauopathy was different – genetic in male and sporadic PSP in the female. In couple #3, both spouses had PD. The onset was separated by 20 years, and the wife had a strong family history of PD while the husband had sporadic PD. Thus, neither the tauopathy (couple #2) nor the PD (couple #5) was transmitted to the partners. These data indicate that neither of the common PS variants – PD, tauopathy, PSP, or MSA, was transmitted from one spouse to the other.
Genetic consideration
Only one patient (male, couple #1) had tauopathy consequent to LRRK2 mutation and strong family history of that, thus excluding genetic basis of conjugal PS. Future genetic developments may add to our knowledge.
Shared environments
In four of the five couples the PS onset was within three years. However if the shared environments produced PS, we would have expected the same disease process in both spouses – that was not the case in these couples. In Couple #3 where both spouses had PD if shared environments was the cause, one would have expected a closer calendar year of onset. We can therefore exclude shared environmental basis of PS. The same conclusion was made by others who studied shared environments in more detail31.
Incidental PS
The final and the most likely explanation, is that these are incidental PS cases that happened to be married to each other. Similar conclusion was reached in another conjugal case report32. Parkinsonism is a common disorder in later age. The age specific incidence of PS rises remarkably with advancing age and the cumulative risk of developing parkinsonism in those 60 years and older is 4%45.
In summary, conjugal PS in non-consanguineous couples is a chance occurrence – it is neither transmitted from one partner to the other nor is it caused by shared environment.
Acknowledgements
We are grateful to Ms. L. Beatty, Dr. S. Akhtar and Miss E. Rajput for their assistance in preparing the manuscript.
The authors have no conflict of interest.
References
- Rajput AH, Rozdilsky B, Rajput A. Accuracy of Clinical Diagnosis in Parkinsonism A Prospective Study. Can J Neurol Sci 1991; 18: 275-278.
- Pagano G, Ferrara N, Brooks DJ, et al. Age at onset and Parkinson disease phenotype. Neurology. 2016; 86: 1400-1407.
- Postuma RB, Berg D, Stern M, et al. MDS Clinical Diagnostic Criteria for Parkinson's disease. Mov Disord. 2015; 30: 1591-1599.
- Reinoso G, Allen JC, Au WL, et al. Clinical evolution of Parkinson's disease and prognostic factors affecting motor progression 9-year follow up study. Eur J Neurol. 2015; 22: 457-463.
- Jellinger K, Marsden CD, Fahn S. The Pathology of Parkinsonism. In eds Movement Disorders 2 London Butterworths and Co. 1987; 124-165.
- Duvoisin RC, Golbe LI. Toward a definition of Parkinson's disease. Neurology 1989; 39:746.
- Bower JH, Dickson DW, Taylor L, et al. Clinical correlates of the pathology underlying parkinsonism. A population perspective Mov Disord. 2002; 17: 910-916.
- Gelb DJ, Oliver E, Gilman S. Diagnostic criteria for Parkinson disease. Arch Neurol. 1999; 56: 33-39.
- Hughes AJ, Daniel SE, Kilford L, et al. Accuracy of clinical diagnosis of idiopathic Parkinson's disease A clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry. 1992; 55: 181-184.
- Adler CH, Beach TG, Hentz JG, et al. Low clinical diagnostic accuracy of early vs advanced Parkinson disdease A clinicopathologic study. Neurology. 2014; 83: 406-412.
- Langston JW, Schule B, Rees L, et al. Multisystem Lewy body disease and the other parkinsonian disorders. Nature Genetics. 2015; 47: 1378-1384.
- Puschmann A. Monogenic Parkinson's disease and parkinsonism Clinical phenotypes and frequencies of known mutations. Parkinsonism Relat Disord. 2013; 19: 407-415.
- Esiri MM, Kennedy, Adams JH. PGE Virus diseases In eds Greenfield's Neuropathology Fifth Edition ed. New York Oxford University Press. 1992; 335-399.
- Duvoisin RC, Yahr MD. Encephalitis and Parkinsonism. Arch Neurol. 1965; 12: 227-239.
- Friedman JH, Stern MB, Koller WC. Postencephalitic Parkinsonism In eds Parkinsonian Syndromes Marcel Dekker Inc. 1993; 203-226.
- Langston JW, Ballard P, Tetrud JW, et al. Chronic Parkinsonism in humans due to a product of Meperidine analog Synthesis. Science. 1983; 219: 979-980.
- Luo C, Rajput AH, Akhtar S, et al. α-synuclein and tyrosine hydroxylase expression in acute rotenone toxicity. Int J Mol Med. 2007; 19: 517-521.
- Shuaib UA, Rajput AH, Robinson CA, et al. Neuroleptic induced parkinsonism: Clinicopathological study. Mov Disord. 2016; 31: 360-365.
- Ahn TB, Langston JW, Aachi VR, et al. Relationship of neighboring tissue and gliosis to α-synuclein pathology in a fetal transplant for Parkinson's disease. Am J Neurodegener Dis. 2012; 1: 49-59.
- Braak H, Del Tredici K. Assessing fetal nerve cell grafts in Parkinson's disease. Nature Medicine. 2008; 14: 483-485.
- Kordower JH, Chu Y, Hauser RA, et al. Lewy body like pathology in long term embryonic nigral transplants in Parkinson's disease. Nature Medicine. 2008; 14: 504-506.
- Li JY, Englund E, Holton JL, et al. Lewy bodies in grafted neurons in subjects with Parkinson's disease suggest host to graft disease propagation. Nature Medicine. 2008; 14: 501-503.
- Brundin P, Li JY, Holton JL, et al. Research in motion the enigma of Parkinson's disease pathology spread. Nat Rev Neurosci. 2008; 9: 741-745.
- Angot E, Steiner JA, Hansen C, et al. Are synucleinopathies prion-like disorders? The Lancet 2010; 9:1128-1138.
- Peelaerts W, Bousset L, Van der Perren A, et al. α-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature. 2015; 522: 340-344.
- Sanders DW, Kaufman SK, DeVos SL, et al. Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron. 2014; 82: 1271-1288.
- Scherfler C, Schwarz J, Antonini A, et al. Role of DAT SPECT in the diagnostic work up of Parkinsonism. Mov Disord. 2007; 22: 1229-1238.
- Stoessl AJ. Neuroimaging in Parkinson's Disease. Neurotherapeutics. 2011; 8: 72-81.
- Ravina B, Eidelberg D, Ahlskog JE, et al. The role of radiotracer imaging in Parkinson disease. Neurology. 2005; 64: 208-215.
- Tolosa E, Borght TV, Moreno E, et al. Accuracy of DaTscan (123I-Ioflupane) SPECT in diagnosis of patients with clinically uncertain parkinsonism 2 year follow up of an Open Label Study. Mov Disord. 2007; 22: 2346-2351.
- Willis AW, Sterling C, Racette BA. Conjugal parkinsonism and Parkinson disease A case series with environmental risk factor analysis. Parkinsonism Relat Disord. 2010; 16: 163-166.
- Strang RR. Conjugal Parkinsonism. Dis Nerv Syst. 1967; 28: 814-815.
- Miwa H, Kondo T. Conjugal parkinsonism Multiple system atrophy and Parkinson's disease. Parkinsonism Relat Disord. 2010; 16: 232.
- Counihan TJ. Conjugal Parkinson's disease. The Lancet. 2003; 361:252.
- Ramani M, Saur DP, Rabin M, et al. Conjugal and familial Lewy body disorders A report of one family. Parkinsonism Relat Disord. 2013; 19:498.
- Rajput AH, Ferguson LW, Robinson CA, et al. Conjugal parkinsonism Clinical pathology and genetic study. No evidence of person to person transmission. Parkinsonism Relat Disord. 2016; 31: 87-90.
- Rajput AH, Rajput A. Saskatchewan Movement Disorders Program. Can J Neurol Sci. 2015; 42: 74-87.
- Rajput AH, Voll A, Rajput ML, et al. Course in Parkinson's Disease subtypes A 39 year clinicopathological study. Neurology. 2009; 73: 206-212.
- Rajput AH, Rozdilsky B, Rajput A, et al. Levodopa efficacy and pathological basis of Parkinson Syndrome. Clin Neuropharmacol. 1990; 13: 553-558.
- Rajput AH, Rajput ML, Robinson CA, et al. Normal substantia nigra patients treated with levodopa Clinical therapeutic and pathological observations. Parkinsonism Relat Disord. 2015; 21: 1232-1237.
- Rajput AH, Sitte H, Rajput A, et al. Globus pallidus dopamine and Parkinson motor subtypes Clinical and brain biochemical correlation. Neurology. 2008; 70: 1403-1410.
- Vilarino-Guell C, Rajput A, Milnerwood AJ, et al. DNAJC13 mutations in Parkinson disease. Hum Mol Genet. 2014; 23: 1794-1801.
- Pifl C, Rajput A, Reither H, et al. Is Parkinson's disease a vesicular dopamine storage disorder Evidence from a study in isolated synaptic vasicles of human and nonhuman primate striatum. J Neurosci. 2014; 34: 8210-8218.
- Paris-Robidas S, Brochu E, Sintes M, et al. Defective dentate nucleus GABA receptors in essential tremor. Brain. 2012; 135: 105-116.
- Elbaz A, Bower JH, Maraganore D, et al. Risk tables for parkinsonism and Parkinson's disease. J Clin Epidemiol. 2002; 55: 25-31.