
Karenitecin (BNP1350) radiosensitizes glioma cells with an associated increase in caspase activation and reactive oxygen species.
Deepika Rajesh, H. Ian Robins* and Steven. P. Howard
Abstract
The prognosis for patients with high-grade glioma remains dismal illustrating the need for the development of chemotherapeutic strategies intended to synergize with treatment modalities of proven efficacy such as radiation therapy. Karenitecin, (BNP1350,7-[(2-trimethylsilyl)ethyl]-20(S)-camptothecin), is a semisynthetic, orally administrable camptothecin, with physiochemical properties favoring increased accumulation of active drug within the central nervous system. This study was initiated to determine whether Karenitecin could potentiate radiosensitivity in T98G and MO59K glioma cell lines. Using clinically relevant doses of each agent, it was demonstrated that Karenitecin pretreatment significantly enhances radiosensitivity and was shown to be synergistic by isobolographic analysis. Karenitecin treatment resulted in a transient arrest in the G2/M phase of the cell cycle with an associated increase in expression of cyclin B1. It was further demonstrated that Karenitecin treatment results in activation of caspases, generation of reactive oxygen species, collapse of the mitochondrial membrane potential and inhibition of total protein kinase C, all of which could enhance radiosensitivity. These in vitro data suggest that Karenitecin has potential as a radiosensitizer in the treatment of malignant glioma.
Introduction
In spite of extensive efforts, minimal improvements in the treatment Glioblastoma Multiforme (GBM) have been realized. Stupp et al1,2 recently reported the results of a Phase III trial demonstrating that patients who were randomized to receive temozolomide (TMZ) during and after radiation therapy (RT) had a significantly better outcome than those patients randomized to receive RT alone. The median survival was increased by two months compared to RT alone with 35% of the patients surviving beyond 18 months representing the most significant advancement the treatment of GBM over the past 30 years. This successful clinical trial serves as “proof of principle” that treatments of known efficacy such as radiotherapy can be delivered in conjunction with chemotherapeutic agents having specific molecular targets such as TMZ to improve treatment outcomes. However, the majority of patients will ultimately succumb to their disease and a substantial percentage of patients will not respond to TMZ, regardless if given in combination with RT or as monotherapy, due to expression of the DNA repair enzyme alkylguanine-DNA alkyltransferase3.
Campothecins, specific inhibitors of topoisomerase I (topo I), are a relatively new class of anticancer agents that have come into widespread clinical use. Topo I relieves excessive DNA supercoiling that occurs during DNA replication by inducing single strand DNA breaks followed by a DNA-rejoining reaction. The relegation step which is catalyzed by topo I is inhibited by the camptothecins resulting in DNA damage and consequently triggering apoptosis4-6. The water-soluble camptothecins, Irinotecan (CPT-11; Camptosar; Pfizer, Inc., New York, NY) and Topotecan (Hycamtin, GlaxoSmithKline, Munich, Germany) have been extensively evaluated as single agents in the treatment of GBM7,8. Response rates as high as 15% have been reported, although most studies have reported more modest results9-12. Both in vitro and in vivo studies have demonstrated that Topotecan has radiosensitizing properties with a dose enhancement ratio of ≥2.0. This radiosensitization appears to be schedule dependent, S phase specific, and not directly related to drug cytotoxicity. The mechanism of the observed synergy between radiation and topotecan results from altered DNA lesion modification and enhanced apoptosis5,13-16. This promising preclinical data was the basis for a phase II study performed by the Radiation Therapy Oncology Group in adult GBM evaluating daily Topotecan as a radiosensitizer7. In addition, the maximum tolerated dose (MTD) of daily Topotecan given during a six week course of radiotherapy in pediatric brain stem glioma patients was determined in the Children’s Cancer Group 0925 Phase I study14. Using this MTD, the French Society of Pediatric Oncology has recently completed a Phase II trial also evaluating Topotecan as a radiosensitizer in pediatric brain stem glioma patients13. Unfortunately, all three of these studies failed to demonstrate a survival benefit.
One of the potential limitations effecting efficacy of the water-soluble topo I inhibitors, such as Irinotecan and Topotecan, is that the less-active, open-ring carboxylate form predominates at physiologic pH. Karenitecin, a semisynthetic camptothecin, (BNP1350, 7-[(2-trimethylsilyl)ethyl]-20(S)-camptothecin produced by BioNumerik Pharmaceuticals, San Antonio, TX), displays increased stability at physiologic pH maintaining a high percentage of drug in the active lactone form enhancing tissue penetration and bio-availability compared to the water-soluble camptothecins12. There is also experimental evidence that the lactone form of the camptothecins binds directly and noncovalently to double-stranded DNA and single-stranded DNA in the absence of Topo I and that this binding may play an additional role in their anticancer activity17. Phase II clinical trials have been recently completed evaluating Karenitecin in patients with non-small cell lung cancer, metastatic melanoma and ovarian cancer18-21. Karenitecin is highly lipophilic favoring blood-brain barrier transit and has demonstrated activity in xenografts derived from childhood and adult high-grade gliomas including sublines resistant to procarbazine and busulfan22. Results of a phase I trial have established the MTD of single agent Karenitecin in adult GBM patients23. We have previously reported the ability of Karenitecin to act as a radiation sensitizer in glioma cell lines24.
In the report to follow, we extend those studies as we mechanistically characterize the interactions of Karenitecin and radiation. Using clinically relevant doses of radiation, we demonstrate that relatively low doses of Karenitecin effectively radiosensitizes glioma cell lines in vitro. Karenitecin treatment initiates the onset of apoptosis resulting in cell cycle arrest, reduction in total Protein Kinase C activity (PKC), and collapse of the mitochondrial membrane potential resulting in the generation of reactive oxygen species with subsequent activation of caspases. All of these cellular processes are augmented by radiation treatment. The in vitro data presented in this report support the use Karenitecin has as a potential radiosensitizer in the treatment of grade 4 glioma.
Experimental Procedures
Materials:
Karenitecin (BNP1350) was provided by Dr. Frederick H. Hausheer; Bionumerik Pharmaceuticals Inc; San Antonio, TX. Antibodies to Cyclin B and D were purchased from Santacruz Biotechnology Santa Cruz, CA. All other chemicals including propidium iodide (PI), RNase H, DMSO, and cycloheximide were purchased from Sigma Chemical Co., St. Louis, MO. The Pan Caspase kit was purchased from CHEMICON International, Inc Temecula, CA. The mitochondrial dyes DCFDA, Mitotracker Green and JC-1 were purchased from Molecular Probes, Inc; Eugene, OR. The non-radioactive PKC assay kit was purchased from EMB Biosciences Inc; La Jolla, CA and the Annexin staining kit was purchased from Clonetech (Palo Alto, CA).
Cell lines:
The T98G25 and MO59K26 cell lines were obtained from ATCC and maintained in a humidified incubator with 5% CO2 at 37°C and grown in DMEM F12 medium containing 10% fetal bovine serum, 1% penicillin, streptomycin and 1mM non-essential amino acids.
Cell treatments and clonogenic survival assays:
Clonogenic survival assays were performed to evaluate the cytotoxic effects of different doses of Karenitecin, radiation and a combination of both agents. A stock solution of Karenitecin in DMSO was used for all experiments. T98G and MO59K cells were treated with varying doses of Karenitecin (0.1nM to 10nM) for a period of 72 hours. Control dishes were treated with medium alone. All cells were irradiated at a dose rate of approximately 4.5 Gy/min using a 137Cs irradiator. The dose of radiation ranged from 1.0 Gy to 8.5 Gy for T98G cells and 1.0 Gy to 5.5 Gy for MO59K cells. Following irradiation, both Karenitecin treated and untreated cells were harvested, washed with PBS, plated at the desired cell number and incubated for two weeks. Colonies were stained with crystal violet and quantified. Survival was determined as the ratio of plating efficiencies for each irradiated group to that of the untreated control.
Isobologram analysis and curve fitting:
Synergy of cooperative cytotoxicity was determined by isobologram analysis as described by Berenbaum27-29. Isobolograms were constructed using an isoeffect of surviving fraction (SF) = 0.2. The survival curves for Karenitecin, radiation and the combination of Karenitecin and radiation, were fitted to the equation SF= 1 - (1 – e-kD)N, which corresponds to the single-hit, multi target model, using the Matlab and Sigma Plot software package. In this equation, SF is the surviving fraction, D is the dose of drug or radiation, and k and N are curve-fitting constants. This equation was also used to fit all survival curves in these experiments to obtain the corresponding k and N values. Using these curve fits, and following the method of Berenbaum27-29 isobolograms were constructed and graphed using the following equation: x/Dx + y/Dy = 1, where Dx is the dosage of the drug alone (in nM) for which SF (drug alone) = 0.2, and Dy is the level of radiation (in Gy) at which SF (radiation alone) = 0.2. Using the single-hit, multi-target model (with the values of k and N calculated as explained above), we then established each (x,y) combination, where x is the drug dosage and y is the dose of radiation, for which the corresponding SF(x of drug, y of radiation) = 0.2. Synergy is observed when x/Dx + y/Dy < 1; antagonism is observed when x/Dx + y/Dy > 1; and lack of interaction is observed when x/Dx + y/Dy = 1. As demonstrated by Berenbaum27-29 this selection criteria eliminates the attribution of false-positive synergistic or additive effects for a sham combination of an agent with itself; a common error made in the literature is to use supposedly “more stringent” criteria that in fact tends to show synergy for sham combinations of an agent with itself.
Flow cytometry:
The percent of cells undergoing apoptosis and cell cycle distribution were quantified as described by Darzynkiewicz et al30. Cells were treated with 0.1nM-10nM Karenitecin for periods of up to 72 hours. At the indicated times after treatment the cells were harvested by trypsin-EDTA and washed twice with ice-cold PBS. The cell pellet was resuspended in PBS, fixed in ice-cold 95% ethanol and stained using a PI staining solution containing 20ug/ml PI, 200ug/ml RNase H and 0.1% NP40, filtered through a 40 µM pore nylon mesh (Tetkop, Inc.) and analyzed using a Beckton-Dickinson FACStar plus flow cytometry with an excitation wave length of 488 nM. The percentage of cells in sub-G0/G1 and the distribution of cells in G0/G1, S and G2/M phases were determined using the Modfit LT version 2.0 (Verity Software, Topham, Maine) and Cell Quest software (Becton Dickinson, San Jose, CA).
Annexin V and PI staining:
Dual staining to detect the presence of annexin V and PI was performed by flow cytometry. Glioma cell lines were treated with varying doses of Karenitecin for the indicated times, harvested using cell dissociation solution, washed and subsequently stained with antibody to annexin V conjugated to fluorescein isothiocynate (FITC) and PI [10µg/ml] using the Apoalert Kit (Clonetech, Palo Alto, CA). Viable [annexin V¯/PI¯], pre-apoptotic [annexin V+/PI¯], and apoptotic [annexin V+ /PI+], and the residual damaged [annexin V¯/PI+] cells were quantified using Cell Quest software (Becton Dickinson, San Jose, CA).
Schematic representation of the regulation of Fas and Faim during the different phases of the neurologic diseases studied. Faim2 is constitutitvely expressed, inhibiting Fas dependent apoptotic cell death. After the acute insult, cell delayed and programmed death occurrs by excitotoxicity, oxidative stress and inflammation. In this phase, constitutive Faim2 can prevent Fas induced cell death, and increasing Faim2 can further reduce cell death. In the last phase, however, down regulation of Faim2 may allow alternative Fas signalling that underlies regenerative processes such as proliferation and differentiation of neuronal precursors and neurite outgrowth.
Mitochondria related event staining:
Karenitecin treated cells with and without subsequent irradiation were harvested using cell dissociation solution and stained for 15 min at 37°C with a 10µg/ml solution 2'7'-dichlorodihydrofluorescein diacetate (DCFH-DA) to detect changes in intracellular reactive oxygen species (ROS) or 10µg/ml solution JC-1 to measure changes in mitochondrial membrane potential (Δψm)22,25-28. Stained cells were washed once and resuspended in ice cold PBS and immediately analyzed by flow cytometry (FACScan, Becton Dickinson, USA). In the presence of ROS, intracellular esterases cleave acetate groups from DCFDA, trapping non-fluorescent 2', 7'-dichlorofluorescein in the cells. Subsequent oxidation yields fluorescent DCF, which detects transient rises in ROS. In healthy cells, JC-1 stains the mitochondria bright red. The negative charge established by the intact mitochondrial membrane potential allows the lipophilic dye to enter the mitochondrial matrix where it accumulates. After exceeding a critical concentration, JC-1 forms aggregates that exhibit red fluorescence at 590 nm. Under conditions where the mitochondrial membrane collapses, JC-1 cannot accumulate within the mitochondria, but remains in the cytoplasm in a green fluorescent monomeric form that emits green fluorescence at 527 nm. The mitochondrial membrane potential was measured by comparing the changes in the green/red fluorescence ratio.
Cyclin Staining:
Cyclins were stained and quantified using established methods31. Sub-confluent cultures of T98G and MO59K cells were treated with varying doses of Karenitecin with and without subsequent irradiation, harvested and fixed in ice-cold ethanol overnight at 40C. Cells were then washed, suspended in permeabilization solution (0.25%v/v Triton X-100 in PBS pH 7.4), kept on ice for 5 minutes, resuspended in 100μl wash buffer (1% BSA in PBS) and incubated with antibodies to cyclins B, D or isotype control for 60 minutes at room temperature. The cells were then washed, and stained with goat anti-mouse FITC for 30 minutes at room temperature and then treated with 5 μg/ml PI in 200μg/ml DNase-free RNase A. Propidim iodide staining was used to determine the cellular distribution throughout the cell cycle that was correlated with cyclin expression by simultaneously staining for cyclin B1 and cyclin D1 with FlTC conjugated antibodies in the G0/G1 and G2/M phases of the cell cycle. Stained cells were analyzed on a FACScan flow cytometer (Becton Dickinson, San Jose, CA) and the results were acquired using CELL quest software and the ModfitLT version 2.0 (Verity Software, Topham, Maine).
Pan-caspase assay:
Karenitecin treated cells with and without subsequent irradiation were assayed for pan-caspase activity using the Pan-Caspase kit (Chemicon, Temecula, CA). Following treatment, cells were washed and incubated with carboxyfluorescein-labeled fluoromethyl ketone peptide inhibitor of caspase (SR-VAD-FMK), for 1 hour at 37°C. The cells were washed and the fluorescence was recorded using a plate set to an excitation wavelength of 550nM and emission of 595nM.
PKC activity:
PKC activity was assayed using non-radioactive methods (EMB Biosciences Inc.). The assay kit utilizes crude cellular extracts and is based on an ELISA method that utilizes a synthetic PKA/PKC pseudosubstrate and a monoclonal antibody recognizing the phosphorylated form of the peptide. T98G and MO59K cells were treated with 10nM Karenitecin at various time points with and without subsequent irradiation, harvested by scraping and cell lysates were made according to the manufacturer’s instructions. 20µg of the of the cell lysate was incubated with component mixture containing phosphtadiyl serine (500µg/ml), 20mM calcium chloride, 1MM ATP, and 20mM EGTA. A parallel set of each sample was incubated in the component mixture lacking calcium and phosphtadiyl serine (500µg/ml). The OD (at 495nM) readings from this set were subtracted from the set containing phosphtadiyl serine and calcium chloride to determine total PKC activity.
PKC isoform western blot analysis:
Crude cell lysates of T98G and MO59K cells were made in buffer containing [50 mM HEPES (pH 7.5), 150 mM NaCl, 10% glycerol, 1% Triton X-100, 1.5 mM MgCl2, 10 µg/ml aprotinin, 10 µg/ml leupeptin, 1 mM phenylmethylsulfonyl fluoride, 200 µM Na3VO4, 200 µM NaF, and 1 mM EGTA and centrifuged at 14,000 g for 30 min at 4°C. The cell lysate was loaded on 12% SDS gels, proteins were transferred to 0.45 µm of Hybond-P polyvinylidene difluoride transfer membrane (Amersham Biosciences, Piscataway, NJ) and incubated with the appropriate primary and secondary antibodies, and the detection signal was developed with Amersham’s enhanced chemiluminescence reagent. Rat brain lysate was used as a positive control.
Results
Karenitecin radiosensitizes glioblastoma cell lines:
The clonogenic survival of T98G and MO59K cell lines pretreated with varying concentrations of Karenitecin (0.1 nM-10 nM) and subsequently irradiated (1.0-8.5Gy) demonstrated a dose dependent sensitization to cell kill by radiation treatment (Figures 1A and 1B). Pretreatment with Karenitecin exposure alone up to a concentration 10nM did not affect cell survival substantially. Higher concentrations of Karenitecin ranging between 20–100nM resulted in a substantial reduction of the surviving fraction and hence radiosensitization experiments were performed with Karenitecin concentrations in a subtoxic range between 0.1-10nM. Radiosensitization was observed at doses as low as 1nM Karenitecin at clinically relevant doses of radiation. To determine whether the effects observed were additive or super-additive (synergistic), isobologram analysis was performed as described by Berenbaum29,32 (Figures 1C and 1D). The graphed line is calculated representing the expected cytotoxicity assuming purely additive interactions between agents; the data points that lay on the left indicate super-additivity, or synergism. If data points were observed lying to the right of the graphed line, this would indicate antagonism. The combination of 1nM Karenitecin + 2.13Gy radiation and 0.1nM Karenitecin + 4.06Gy radiation were synergistic in T98G cells. Similarly, 0.1nM Karenitecin + 2.63Gy radiation and 1nM Karenitecin + 1.82Gy radiation were synergistic in MO59K cells.
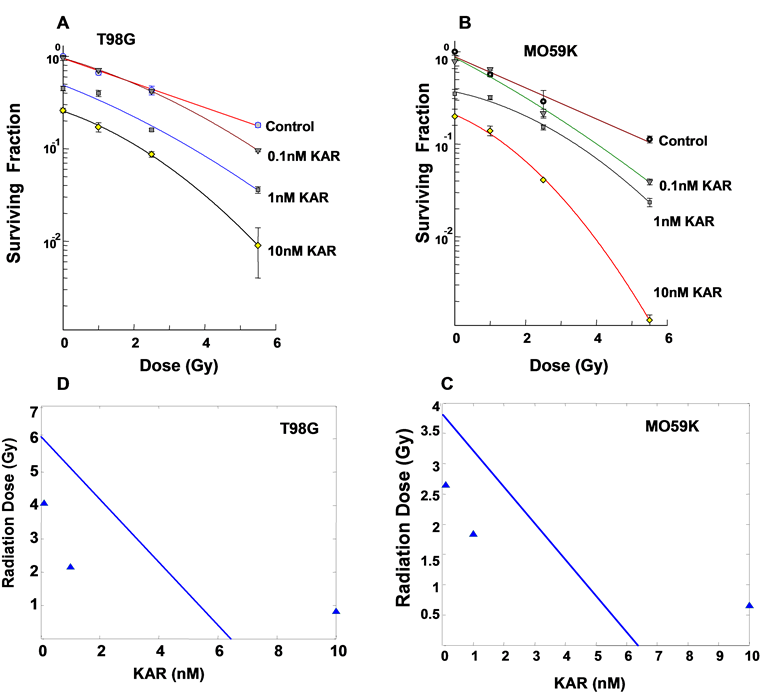
Figure 1: Karenitecin pretreatment induces sensitivity to cell death via radiation; Subconfluent cultures of T98G cells (A) and MO59K (B) were treated with 0.1nM, 1nM and 10nM concentrations of Karenitecin for 72 hours and irradiated (1.0-8.5 Gy). Control cultures were treated with the indicated doses of radiation, or Karenitecin or vehicle (DMSO) alone, harvested and plated at clonal densities and allowed to grow for 14 days. The resulting colonies were stained and counted. Each graphed point represents mean values ± SE values of triplicate dishes (variation less than 5%). Isobologram analysis of T98G (C) and MO59K (D) graphically illustrating cytotoxic interactions between Karenitecin and radiation. The solid line is calculated representing concentrations of both agents required to inhibit cell growth to 80% (survival 0.2) and indicates purely additive interactions. The data points located below the line indicate synergistic interactions, while the points over the envelope indicate antagonistic interaction.
Treatment of cells with 10nM Karenitecin with radiation was not synergistic. These results illustrate that pretreatment with Karenitecin concentrations having only modest toxicity effectively radiosensitizes glioma cell lines at clinically relevant doses of radiation.
Karenitecin induced apoptosis and activation of caspases:
Topo I inhibitors elicit cytotoxicity by inducing apoptosis4. Hence, the ability of Karenitecin to initiate the apoptotic pathway in glioma cell lines was evaluated. Treatment of T98G and MO59K cells with Karenitecin resulted in a dose and time dependent increase in the percent of cells in subG0/G1 indicating the onset of apoptosis (Figures 2A and 2B). Karenitecin induced cell death was confirmed by staining for the presence of Annexin V by flow cytometry. Karenitecin treated cells demonstrated an increase the early apoptotic [annexin V+/PI¯] and apoptotic [annexin V+/PI+] populations of cells (Figure 2C). The central component of apoptotic process involves the activation of caspases33. Karenitecin treated T98G and MO59K cells were assayed for activation of caspases-3 (Figure 2D). Karenitecin treatment results in activation of zVADfmk- and yVADcmk-sensitive caspases in both cell lines.
Figure 2: Karenitecin treatment increases the proportion of Sub G0/G1 (apoptotic) cells. T98G cells (A) and MO59K cells (B) were treated with either vehicle (DMSO) or with indicated concentration of Karenitecin. Apoptosis was quantified at 24, 48 and 72 hours after treatment by determining the sub-G0/G1 fraction by flow cytometry. Each data point is an average of duplicate dishes (variation <5%). C: Live cell surface staining of cells undergoing apoptosis by performing staining for (PI and Annexin V) by flow cytometry. Cell lines were treated with varying doses of Karenitecin for 72 hours, harvested, washed with PBS and subsequently stained with antibody to annexin V conjugated to FITC and with PI (10µg/ml). Viable cells [annexin V¯/PI¯], pre-apoptotic cells [annexin V+/PI¯], apoptotic cells [annexin V+/PI+], and residual damaged [annexin V¯/PI+] cells were quantified by flow cytometric analysis. D: Pan Capase activity was determined in T98G and MO59K cells treated with 1, 10 and 100nM Karenitecin for 48 hours, washed and incubated with carboxyfluorescein-labeled fluoromethyl ketone peptide inhibitor of caspase (SR-VAD-FMK) for 1 hour at 37C. Fluorescence was recorded using a plate reader set to an excitation wavelength of 550nM and emission of 595nM. Each value represents mean values ± SE values of triplicate dishes (variation less than 5%).
Karenitecin induced mitochondrial related events:
Mitochondria have been identified as the central processing organelles in most apoptotic pathways. Apoptotic signals from either outside or inside the cell converge in mitochondria. These organelles are thought to sense stress signals and initiate the execution phase of apoptosis34-36. To determine if Karenitecin mediated apoptosis required the participation of mitochondrial dependent changes, we directly measured the depolarization of mitochondrial membrane and generation of reactive oxygen species by using mitochondria specific dyes and flow cytometry. At the initiation of the apoptotic process the electrochemical gradient across the mitochondrial membrane collapses. Loss of the mitochondrial membrane potential (Δψm) is a hallmark for apoptosis that can be measured by detecting the fluorescence of a unique dye, JC-1 (5,5',6,6'-tetrachloro-1,1',3,3' tetraethylbenzimidazolylcarbocyanine iodide/chloride). In viable, non-apoptotic cells, JC-1 accumulates as aggregates in the mitochondria, resulting in red fluorescence. In apoptotic cells, the mitochondrial membrane collapses and JC-1 disperses throughout the cell, maintaining its green fluorescent monomeric form. JC-1 is more advantageous over rhodamines and other carbocyanines, capable of entering selectively into mitochondria, since it causes the fluorescence emission shift from green (~525 nm) to red (~590 nm). Mitochondrial depolarization is indicated by a decrease in the red/green fluorescence intensity ratio. Karenitecin treatment caused a dose dependent increase in the green fluorescence emission in both the cell lines suggesting a collapse of the mitochondrial membrane potential (Figure 3A).
Reactive oxygen species (ROS), including superoxide radical, hydrogen peroxide, hydroxyl radical and singlet oxygen, are continuously generated from the mitochondrial respiratory chain, have powerful oxidative potential, and are known to be capable of resulting in oxidative damage. Endogenous ROS formation and lipid peroxidation result in eventual loss of mitochondrial membrane potential. Many pro-apoptotic signals can raise the cellular ROS level. In addition, the cellular ROS level may be associated with the susceptibility of tumor cells to pro-apoptotic signals35,37. The cell-permeable fluorogenic probe 2’7’-dichlordihydrofluorescin diacetate (DCF-DA) was used to measure oxidative stress. In the presence of ROS, DCFH rapidly undergoes one-electron oxidation to the highly fluorescent compound dichlorofluorescein (DCF).
Radiation, Karenitecin and a combination of these two agents all result in an increase in the generation of ROS (Figure 3B). Pretreatment of cell cultures with Karenitecin prior to irradiation results in a greater increase in ROS than treatment with either agent alone. The increase in ROS observed after Karenitecin exposure could potentially sensitize to other proapoptotic signals such as those generated after irradiation resulting in collapse of the mitochondrial membrane potential and subsequent activation of caspases.
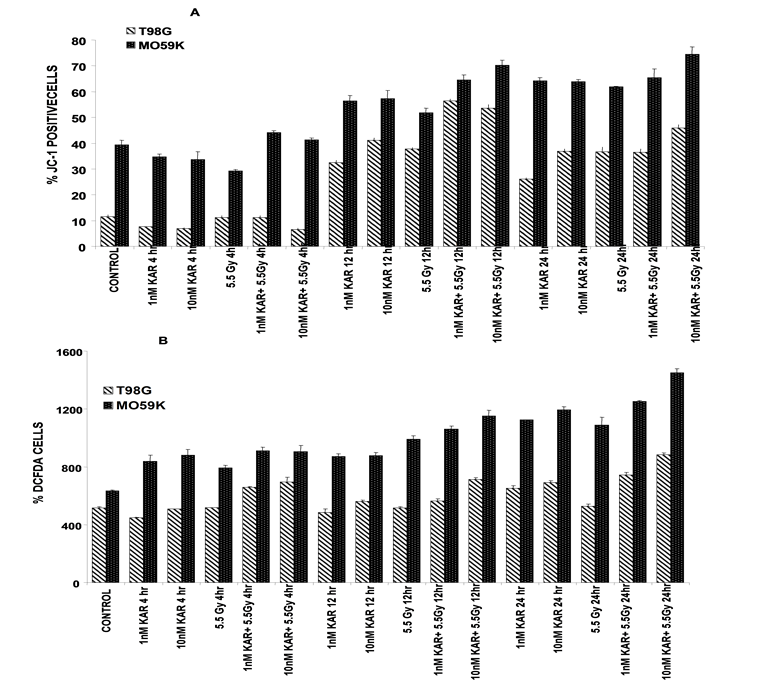
Figure 3: Karenitecin effect on percentage JC-1 cells and % DCFDA positive cells as a function of time and concentration; One of the distinctive feature of the early stages of apoptosis is the disruption of active mitochondria membrane potential and alterations to redox potential. JC-1(5,5’,6,6’-tetrachloro-1,1’,3,3’-tetraethylbenzimidazolylcarbocyanine iodide) is a novel cationic carbocyanine dye. In healthy cells, JC-1 selectively accumulates in mitochondria and exhibits red fluorescence. In apoptotic cells with reduced mitochondria membrane potential, JC-1 exists as a monomer resulting in green fluorescence. (A). Glioma cell lines were treated with the indicated doses of Karenitecin with and without subsequent irradiation, harvested, washed and incubated with JC-1 and then washed with phosphate-buffered saline (PBS) and analyzed on a flow cytometer using 488 nm excitation with 530 nm and 590 nm bandpass emission filters via flow cytometry. The intensity of the green fluorescence was quantified which in turn indicates collapse of the membrane potential ?Ψm. Each value represents mean values ± SE values of triplicate dishes (variation less than 5%).
Karenitecin alters cell cycle distribution and cyclin levels:
Treatment of T98G and MO59K cells with varying concentrations of Karenitecin with or without subsequent radiation treatment resulted in a dose dependent accumulation of cells in the G2/M phase of the cell cycle with a corresponding decrease of the proportion of cells in G0/G1 (Figure 4). Progression through the cell cycle is regulated by the complex interactions between the cyclins (A-E) and the cyclin-dependent kinases (cdks). Distinct cyclins perform different tasks in specific phases of the cell cycle by binding to and activating the appropriate cyclin dependent kinases. Cyclin B promotes G2/M transition, while the D-type cyclins (cyclin D1, D2, and D3) promote cell cycle progression from G1 to S phase38.
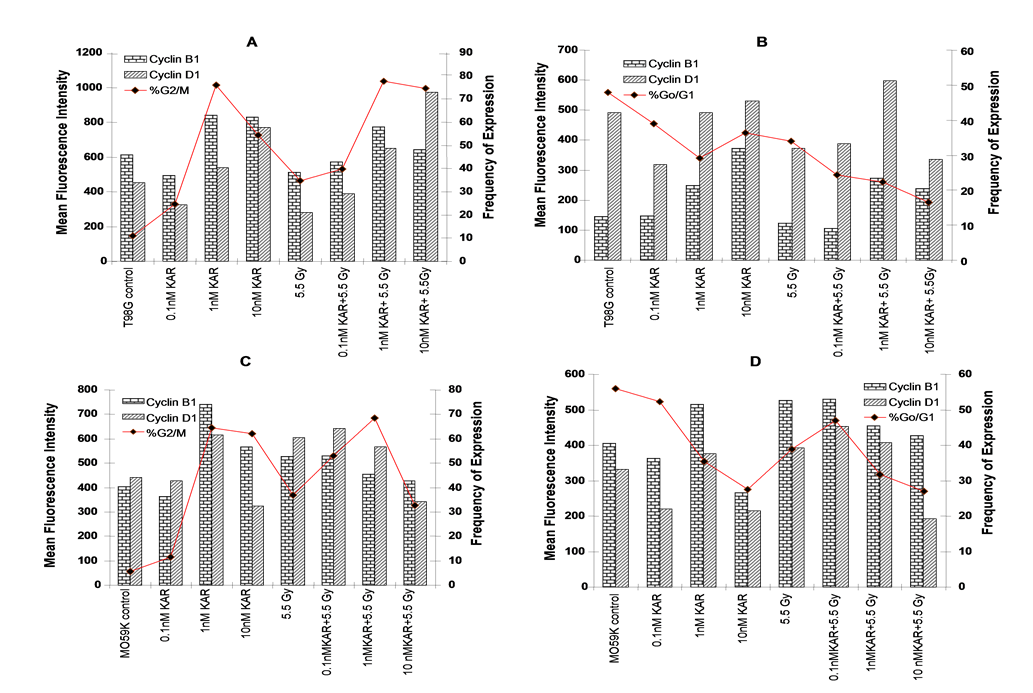
Figures 4: Karenitecin treatment alters cell cycle distribution; Sub-confluent cultures of T98G cells (Figure A and B) and MO59K cells (Figure C and D) and were treated with the indicated concentrations of Karenitecin for 72 hours with and without subsequent irradiation with 5.5 Gy. Control cultures were treated with vehicle (DMSO) alone. Cells were harvested fixed in ethanol, permeabilized and stained using specific antibodies for cyclin D1 or cyclin B1 or the relevant isotype control followed by staining with goat anti-mouse FITC. The antibody stained cells were then stained with PI and analyzed on a FACScan flow cytometer (Becton Dickinson, San Jose, CA). Simultaneous bivariate analysis of DNA content and cyclin expression was performed by staining cells with PI to determine the cell cycle distribution and cyclin staining within the G0/G1 and G2/M phase of the cell cycle was quantified using the cell quest program. The Mean Fluorescence Intensity (MFI) is graphed for cyclin B1 and cyclin D1 staining after subtracting the MFI of the isotype control.
Since Karenitecin treatment resulted in an accumulation of cells in G2/M with a corresponding decrease in G0/G1, we evaluated the expression of cyclin D1 and B1 in the G0/G1 and G2/M phases of the cell cycle by flow cytometry. An increase in the levels of cyclin B1 predominantly in the G2/M phase of the cell cycle was observed correlating with the increase in the percentage of cells in the G2/M phase of the cell cycle following Karenitecin treatment in both cell lines (Figure 4A and 4C). Correlating with the observed decrease in the percentage of cells in the G0/G1 phase of the cell cycle, no significant change or a slight decrease in the levels of cylcin D was detected in cells in G0/G1 (Figure 4B and 4D). In addition, a slight increase in the expression of cyclin B1 was seen in the G0/G1 phase of the cell cycle and an increase in the expression of cyclin D was seen in the G2/M phase of the cell cycle, which could potentially be attributed to unscheduled cyclin production.
Karenitecin treatment alters the levels of PKC:
The role of PKC in glioma tumorigenesis is not well defined. PKC, a family of phospholipid-dependent serine-threonine kinases is comprised of at least 10 isoforms that are divided into the classic PKCs (α, ß1, ß2, and γ ), novel PKCs (δ, ε, η, and θ), and atypical PKCs (PKC ζ and PKC ι/λ;).
The novel and atypical PKC isoforms have been reported to undergo caspase-dependent cleavage in response to various apoptotic stimuli and the accumulation of their constitutively active catalytic fragments has been associated with the regulation of apoptosis39. Recently there have been a number of reports demonstrating synergistic actions of topo I inhibitors and PKC inhibitors suggesting a potentially useful drug combination40-42.
We therefore evaluated the effects of Karenitecin and radiation on PKC activity. Additionally, T98G and M059K glioma cell lines were screened for PKC isoforms using western blotting. Both cell lines expressed most of the PKC isoforms (Figure 5B). Karenitecin treatment of T98G and M059K cells with or without subsequent irradiation resulted in a decrease in total PKC activity (Figure 5A) as reflected by relative activity (i.e., OD at 495nM compared to control). Given the multifunctional role of PKCs in tumor biology, the inhibition of PKC function could interfere with signal transduction pathways critical for survival and contribute to the radiosensitizing effects of Karenitecin.
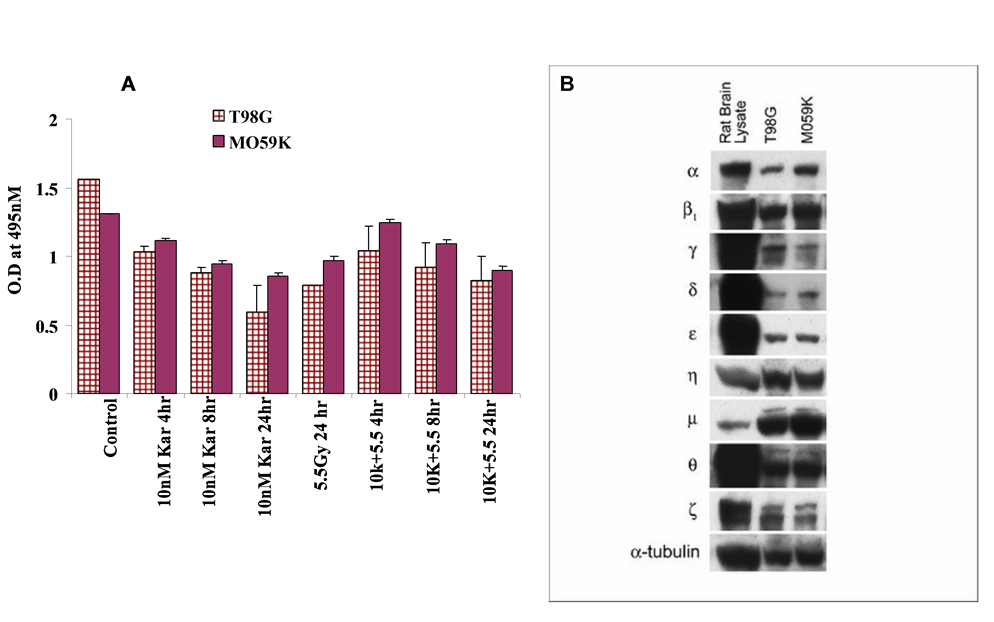
Figure 5: PKC Activity and western blotting PKC isoforms in glioma cell lines; Relative PKC activity as reflected by OD (495nM) in Karenitecin treated cells was assayed using non-radioactive methods. T98G and MO59K cells were treated with 10nM Karenitecin at various time points with and without subsequent irradiation, harvested by scraping and cell lysates made. The PKC activity in the crude cell lysates was assayed using the non-radioactive PKC kit (EMD biosciences) according to the manufacturer’s instructions (A). Cell lysates were made from sub-confluent cultures of T98G and MO59K cells, run on 12% SDS gels, transferred to a PVDF membrane, incubated with PKC isoform specific antibodies, and developed using Amersham’s enhanced chemiluminescence reagent. Rat brain lysate was used as a positive control for these experiments (B).
Discussion
In the present study we demonstrate that the topo I inhibitor Karenitecin is an effective radio sensitizer of T98G and MO59K glioma cell lines. The observed radiosensitization was dose dependent at clinically relevant doses of both Karenitecin and radiation. Using cell death as an endpoint, isobologram analysis illustrates that combinations of Karenitecin and radiation can be synergistic. In Figure 1 (C & D), the line represents the locus of all combinations that would have produced the effect: if there had been no interaction, it is referred to as the zero interaction isobole. All possible dose combinations exhibiting synergy is represented graphically as the area to the left of the zero interaction isobole. For T98G cells, our isobologram analysis predicts for synergy at doses of radiation up to although no greater than 6.0 Gy corresponding to dose of Karenitecin as determined from the analysis of up to although no greater than 6.0 nM. Likewise, for MO59K cells, our isobologram analysis predicts for synergy at doses of radiation up to although no greater than 3.8 Gy corresponding to dose of Karenitecin as determined from the analysis of up to although no greater than 6.3 nM. This information has potential clinical implications in establishing the doses of drug and radiation to be used in combined modality treatments such that therapeutic efficacy is maximized simultaneously minimizing toxicity.
The water-soluble camptothecins Irinotecan and Topotecan elicit cytotoxicity by initiating apoptosis4,43. We verify here, that water-insoluble lipophilic Karenitecin, also elicits cytotoxicity by initiating the apoptotic process (Figure 2 A, B, &C) leading to caspase activation (figure 2D) and subsequent cell death in these glioma cell lines. Apoptotic signals originating from either outside or inside the cell converge in the mitochondria and initiate the execution phase of apoptosis35. At the initiation of the apoptotic process the electrochemical gradient across the mitochondrial membrane collapses resulting in loss of the membrane potential. Using the fluorescent dye JC-1, we directly measured the change in mitochondrial membrane potential in T98G and MO59K cells over time after irradiation, Karenitecin treatment and a combination of these agents (figure 3). Collapse of the mitochondrial membrane with loss of membrane potential was noted 12 hours after treatment with the individual agents as well as the combination. Endogenous ROS formation and lipid peroxidation results in eventual loss of mitochondrial membrane potential. Many pro-apoptotic signals can raise the cellular ROS level. In addition, an elevation in the level of intracellular ROS level may be associated with an increased susceptibility of tumor cells to other pro-apoptotic signals35,37. Radiation, Karenitecin and a combination of these two agents all result in an increase in the generation of ROS. Pretreatment of cell cultures with Karenitecin prior to irradiation results in a greater increase in ROS than treatment with either agent alone. Karenitecin induced apoptosis was accompanied by generation of ROS, collapse of the mitochondrial membrane potential and activation of caspases. The increase in ROS observed after Karenitecin exposure could potentially sensitize the cells to other proapoptotic signals such as those generated after irradiation.
The production of Reactive Oxygen Species (ROS) after Karenitecin treatment was determined using oxidation-sensitive fluorescent probes DCFH-DA (B). Glioma cell lines were treated with the indicated doses of Karenitecin with and without subsequent irradiation, harvested, washed and incubated with 1.0 µM DCFH-DA for 15 min, at 37°C. Cells were washed and analyzed using flow cytometry.
Dead cells and debris were excluded from the analysis by gating on live cells. Data were acquired and analyzed using CellQuest software (Becton-Dickinson, Menlo Park, CA). Each value represents mean values ± SE values of triplicate dishes (variation less than 5%).
Irradiation of MO59K cells results in G2/M arrest with a corresponding increase in cyclin B1 and cyclin D1 levels in both G0/G1 and G2/M compartments. Irradiation of T98G cells also results in G2/M arrest, although, little or no change in cyclin B1 levels and decreased cyclin D1 levels are observed in both G0/G1 and G2/M compartments. T98G and MO59K cells treated with both Karenitecin and radiation expressed cyclin B1 and D1 in the G0/G1 and G2/M cell cycle compartments qualitatively similar to cultures treated with Karenitecin only.
Increase in the levels of Cyclin D1 levels were observed in the G2/M compartments of both cell lines in the presence of Karenitecin and radiation. Irradiation of T98G cells resulted in a G2/M arrest, with no change in cyclin B1 levels. Conversely, irradiation of MO59K cells also results in G2/M arrest with a corresponding increase in cyclin B1 levels in both G0/G1 and G2/M compartments. MO59K cells revealed an increase in cyclin D1 levels in the G0/G1 compartment following treatment with 0.1nM and 1nM Karenitecin and radiation while T98G revealed the same effect following treatment with 1nM Karenitecin and radiation. Both cell lines revealed increase in cyclin D1 levels in the G2/M compartment of the cell cycle following pretreatment of Karenitecin and radiation.
Karenitecin treatment has been reported to up-regulate cyclin B1/cdc2 protein expression with a corresponding increase of phosphorylation of the G2/M checkpoint kinase, chk1, resulting in G2/M arrest of HCT-8 colon cancer cells44. In our study, Karenitecin treatment increased cyclin D1 levels. The significance of this observation is not entirely clear. However, by increasing cyclin D1 protein levels, CDK4 kinase activity is increased disrupting p53-independent initiation of G1 arrest. Abrogation of the degradation of cyclin D1 has been shown to sensitize cells to genotoxic stress including radiation45. In addition, over expression of cyclin D1 has been demonstrated to increase radio sensitivity of squamous carcinoma cells and breast cancer cells by modulating the radiation-induced G2/M transition46. This redistribution of cell cycle associated proteins would favor the accumulation of cells in the radiosensitive G2/M compartment. Because > 80% of GBMs have alterations in the p16INK4a cyclin D1-Rb pathway leading to the over expression of the CDK4 and CDK6 genes, Karenitecin may prove to be a particularly effective radiosensitizing agent in the treatment of GBM47,48.
PKC is implicated in regulation of cell cycle progression, induction of apoptosis, and sensitivity to radiation. PKC η contributes to survival of erythroleukemia cells after irradiation49, while a reduction in PKC ζ activity in glioma cells results in increased sensitivity to UV irradiation50. Camptothecin treatment has been reported to increase activity of PKC α and PKC δ in HL60 cells51. We demonstrate here that T98G and MO59K glioma cell lines express most of the PKC isoforms (figure 5A) and Karenitecin treatment results in a decrease in total PKC activity (Figure 5B; a typical western blot) in both cell lines. The inhibition of PKC activity was not altered by subsequent radiation treatment52. Karenitecin induced inhibition of PKC activity prior to radiation treatment could enhance radiation induced DNA damage and attenuate the repair of damaged DNA in glioma cells. This effect has been demonstrated in C6 glioma cells treated with the PKC inhibitor Staurosporine52. Inhibition of PKC activity could interfere with signal transduction pathways critical for survival and contribute to the radio sensitizing effects of Karenitecin.
In summary, Karenitecin is an orally administrable novel camptothecin, which crosses the blood brain barrier. In a clinical scenario, it could easily be administered during a course of radiotherapy. We have demonstrated that Karenitecin is an effective radio sensitizer of glioma cell lines using clinically relevant doses of both Karenitecin and radiation. Karenitecin treatment results in the accumulation of cells in the G2/M compartment of the cell cycle, activation of caspases, generation of ROS, collapse of the mitochondrial membrane potential and inhibition of PKC, all of which could enhance radio-sensitivity. The in vitro data presented taken collectively suggest that Karenitecin has potential as a radiosensitizer in the treatment of GBM.
Acknowledgements
We thank Dr. Frederick H. Hausheer, Bionumerik Pharmaceuticals Inc, (San Antonio TX); for providing us Karenitecin used in the present study. Funded in part by: Kathleen Reader Neuro-Oncology Fund; Human Oncology Neuro-Oncology Fund; P30 CA014520 (Core Grant, University of Wisconsin Carbone Cancer Center. Flow cytometry provided by the University of Wisconsin Carbone Cancer Center grant P30 CA014520.
References
- Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005; 352: 987-96.
- Stupp R, Weber DC. The role of radio and chemotherapy in glioblastoma. Onkologie. 2005; 28: 315-7.
- Baer JC, Freeman AA, Newlands ES, et al. Depletion of O6-alkylguanine DNA alkyltransferase correlates with potentiation of temozolomide and CCNU toxicity in human tumour cells. Br J Cancer. 1993; 67: 1299-302.
- Sordet O, Khan QA, Kohn KW, et al. Apoptosis induced by topoisomerase inhibitors. Curr Med Chem Anti-Canc Agents. 2003; 3: 271-90.
- Chen AY, Choy H, Rothenberg ML. DNA topoisomerase I-targeting drugs as radiation sensitizers. Oncology (Williston Park). 1999; 13: 39-46.
- Chen AY, Chou R, Shih SJ, et al. Enhancement of radiotherapy with DNA topoisomerase I-targeted drugs. Crit Rev Oncol Hematol. 2004; 50: 111-9.
- Fisher B, Won M, Macdonald D, et al. Phase II study of topotecan plus cranial radiation for glioblastoma multiforme results of Radiation Therapy Oncology Group 9513. Int J Radiat Oncol Biol Phys. 2002; 53: 980-6.
- Ohneseit PA, Prager D, Kehlbach R, et al. Cell cycle effects of topotecan alone and in combination with irradiation. Radiother Onco.l 2005; 75: 237-45.
- Reardon DA, Quinn JA, Rich JN, et al. Phase I trial of irinotecan plus temozolomide in adults with recurrent malignant glioma. Cancer. 2005; 104: 1478-86.
- Reardon DA, Quinn JA, Vredenburgh J, et al. Phase II trial of irinotecan plus celecoxib in adults with recurrent malignant glioma. Cancer. 2005; 103: 329-38.
- Xu Y, Villalona Calero MA. Irinotecan mechanisms of tumor resistance and novel strategies for modulating its activity. Ann Oncol. 2002; 13: 1841-51.
- Garcia Carbonero R, Supko JG. Current perspectives on the clinical experience pharmacology and continued development of the camptothecins. Clin Cancer Res. 2002; 8: 641-61.
- Bernier Chastagner V, Grill J, Doz F, et al. Topotecan as a radiosensitizer in the treatment of children with malignant diffuse brainstem gliomas results of a French Society of Paediatric Oncology Phase II Study. Cancer. 2005; 104: 2792-7.
- Sanghavi SN, Needle MN, Krailo MD, et al. A phase I study of topotecan as a radiosensitizer for brainstem glioma of childhood first report of the Children's Cancer Group-0952. Neuro-oncol. 2003; 5: 8-13.
- Blaney SM, Phillips PC, Packer RJ, et al. Phase II evaluation of topotecan for pediatric central nervous system tumors. Cancer. 1996; 78: 527-31.
- Burch PA, Bernath AM, Cascino TL, et al. A North Central Cancer Treatment Group phase II trial of topotecan in relapsed gliomas. Invest New Drugs. 2000; 18: 275-80.
- Yao S, Murali D, Seetharamulu P, et al. Topotecan lactone selectively binds to double and single stranded DNA in the absence of topoisomerase I. Cancer Res. 1998; 58: 3782-6.
- Daud A, Valkov N, Centeno B, et al. Phase II trial of karenitecin in patients with malignant melanoma clinical and translational study. Clin Cancer Res. 2005; 11: 3009-16.
- Smith JA, Newman RA, Hausheer FH, et al. Evaluation of in vitro drug interactions with karenitecin a novel highly lipophilic camptothecin derivative in phase II clinical development. J Clin Pharmacol. 2003; 43: 1008-14.
- Van Hattum AH, Schluper HM, Hausheer FH, et al. Novel camptothecin derivative BNP1350 in experimental human ovarian cancer determination of efficacy and possible mechanisms of resistance. Int J Cancer. 2002; 100: 22-9.
- Miller AA, Herndon JE, Green MR, et al. Phase II trial of karenitecin in patients with relapsed or refractory non-small cell lung cancer (CALGB 30004). Lung Cancer. 2005; 48: 399-407.
- Keir ST, Hausheer F, Lawless AA, et al. Therapeutic activity of 7-[(2-trimethylsilyl)ethyl)]-20 (S)-camptothecin against central nervous system tumor-derived xenografts in athymic mice. Cancer Chemother Pharmacol. 2001; 48: 83-7.
- Grossman SA, Carson KA, Phuphanich S, et al. New Approaches to Brain Tumor Therapy CNSC. Phase I and pharmacokinetic study of karenitecin in patients with recurrent malignant gliomas. Neuro Oncol. 2008; 10: 608-16.
- Rajesh D, Robins HI, Howard SP. Karenitecin (BNP1350) and Flavopridol as radiosensitizers in malignant glioma. J Neurol Neuromed. 2016; 1: 1-10.
- Stein GH. T98G an anchorage independent human tumor cell line that exhibits stationary phase G1 arrest in vitro. J Cell Physiol. 1979; 99: 43-54.
- Wang J, Hu L, Allalunis-Turner MJ, et al. Radiation induced damage in two human glioma cell lines as measured by the nucleoid assay. Anticancer Res. 1997; 17: 4615-8.
- Berenbaum MC. A method for testing for synergy with any number of agents. J Infect Dis. 1978; 137: 122-30.
- Berenbaum MC. Correlations between methods for measurement of synergy. J Infect Dis. 1980; 142: 476-80.
- Berenbaum MC. What is synergy. Pharmacol Rev .1989; 41: 93-141.
- Darzynkiewicz Z, Bedner E, Smolewski P. Flow cytometry in analysis of cell cycle and apoptosis. Semin Hematol. 2001; 38: 179-93.
- Darzynkiewicz Z, Gong J, Juan G, et al. Cytometry of cyclin proteins. Cytometry. 1996; 25: 1-13.
- Berenbaum MC. Synergy assessment with growth curves. J Infect Dis. 1984; 150: 304.
- Thornberry NA. Caspases key mediators of apoptosis. Chem Biol. 1998; 5: R97-103.
- Granville DJ, Gottlieb RA. Mitochondria regulators of cell death and survival. ScientificWorldJournal. 2002; 2: 1569-78.
- Newmeyer DD, Ferguson Miller S. Mitochondria releasing power for life and unleashing the machineries of death. Cell. 2003; 112: 481-90.
- Ravagnan L, Roumier T, Kroemer G. Mitochondria the killer organelles and their weapons. J Cell Physiol. 2002; 192: 131-7.
- Ueda S, Masutani H, Nakamura H, et al. Redox control of cell death. Antioxid Redox Signal. 2002; 4: 405-14.
- Grana X, Reddy EP. Cell cycle control in mammalian cells role of cyclins cyclin dependent kinases (CDKs) growth suppressor genes and cyclin dependent kinase inhibitors (CKIs). Oncogene. 1995; 11: 211-9.
- Hofmann J. Protein kinase C isozymes as potential targets for anticancer therapy. Curr Cancer Drug Targets. 2004; 4: 125-46.
- Bredel M, Pollack IF, Freund JM, et al. Protein kinase C inhibition by UCN-01 induces apoptosis in human glioma cells in a time-dependent fashion. J Neurooncol. 1999; 41: 9-20.
- Tortora G, Ciardiello F. Antisense strategies targeting protein kinase C preclinical and clinical development. Semin Oncol. 2003; 30: 26-31.
- Chen TC, Su S, Fry D, et al. Combination therapy with irinotecan and protein kinase C inhibitors in malignant glioma. Cancer. 2003; 97: 2363-73.
- Sordet O, Khan QA, Pommier Y. Apoptotic topoisomerase I-DNA complexes induced by oxygen radicals and mitochondrial dysfunction. Cell Cycle. 2004; 3: 1095-7.
- Yin MB, Guo B, Vanhoefer U, et al. Characterization of protein kinase chk1 essential for the cell cycle checkpoint after exposure of human head and neck carcinoma A253 cells to a novel topoisomerase I inhibitor BNP1350. Mol Pharmacol. 2000; 57: 453-9.
- Agami R, Bernards R. Distinct initiation and maintenance mechanisms cooperate to induce G1 cell cycle arrest in response to DNA damage. Cell. 2000; 102: 55-66.
- Coco Martin JM, Balkenende A, Verschoor T, et al. Cyclin D1 overexpression enhances radiation induced apoptosis and radiosensitivity in a breast tumor cell line. Cancer Res. 1999; 59: 1134-40.
- Chakravarti A, Delaney MA, Noll E, et al. Prognostic and pathologic significance of quantitative protein expression profiling in human gliomas. Clin Cancer Res. 2001; 7: 2387-95.
- Buschges R, Weber RG, Actor B, et al. Amplification and expression of cyclin D genes CCND1, CCND2 and CCND3 in human malignant gliomas. Brain Pathol. 1999; 9: 435-42; discussion 2-3.
- Cataldi A, Centurione L, Di Pietro R, et al. Protein kinase C zeta nuclear translocation mediates the occurrence of radioresistance in friend erythroleukemia cells. J Cell Biochem. 2003; 88: 144-51.
- Hussaini IM, Carpenter JE, Redpath GT, et al. Protein kinase C-eta regulates resistance to UV- and gamma-irradiation-induced apoptosis in glioblastoma cells by preventing caspase-9 activation. Neuro-oncol. 2002; 4: 9-21.
- Shimizu T, Cao CX, Shao RG, et al. phosphorylation by protein kinase calpha and proteolysis during apoptosis in human leukemia HL60 cells. J Biol Chem. 1998; 273: 8669-74.
- Zhang W, Yamada H, Sakai N, et al. Sensitization of C6 glioma cells to radiation by staurosporine a potent protein kinase C inhibitor. J Neurooncol. 1993; 15: 1-7.