
Recent advances in modelling of cerebellar ataxia using induced pluripotent stem cells
Maggie M. K. Wong1, Lauren M. Watson1 and Esther B. E. Becker1
Abstract
The cerebellar ataxias are a group of incurable brain disorders that are caused primarily by the progressive dysfunction and degeneration of cerebellar Purkinje cells. The lack of reliable disease models for the heterogeneous ataxias has hindered the understanding of the underlying pathogenic mechanisms as well as the development of effective therapies for these devastating diseases. Recent advances in the field of induced pluripotent stem cell (iPSC) technology offer new possibilities to better understand and potentially reverse disease pathology. Given the neurodevelopmental phenotypes observed in several types of ataxias, iPSC-based models have the potential to provide significant insights into disease progression, as well as opportunities for the development of early intervention therapies. To date, however, very few studies have successfully used iPSC-derived cells to cerebellar ataxias. In this review, we focus on recent breakthroughs in generating human iPSC-derived Purkinje cells. We also highlight the future challenges that will need to be addressed in order to fully exploit these models for the modelling of the molecular mechanisms underlying cerebellar ataxias and the development of effective therapeutics.
Cerebellar ataxias
The cerebellar ataxias are a clinically and genetically diverse group of neurological disorders that primarily affect the cerebellum. Characterized by a progressive loss of motor coordination, other symptoms include dysarthria and disturbance of eye movements1. Cerebellar ataxias may be acquired or inherited, with hereditary ataxias classified into autosomal dominant, autosomal recessive, X-linked or mitochondrial forms. Over 60 genetic subtypes have been identified to date2-4. Among these, the autosomal recessive disorders Friedreich ataxia (FRDA) and ataxia-telangiectasia (A-T), as well as the autosomal dominant polyglutamine spinocerebellar ataxias (SCAs) are the most studied forms of ataxias. Despite significant clinical and genetic heterogeneity, emerging evidence points to the existence of common pathogenic mechanisms that may be shared by several genetically distinct forms of cerebellar ataxias (reviewed in5-8). However, it is still unclear how the proposed pathological pathways ultimately result in cerebellar dysfunction and degeneration, predominantly affecting Purkinje cells.
Understanding disease mechanisms is key to treating neurodegenerative disorders. The heterogeneous nature of the cerebellar ataxias combined with the unavailability of human brain tissue and the lack of reliable disease models have, however, hampered our understanding of the molecular disease mechanisms underlying cerebellar ataxias and thus, the development of effective therapies. Although mouse models of several cerebellar ataxias, including FRDA and SCAs, have provided valuable insights into the pathophysiology of these disorders (reviewed in9), many questions remain about the observed species differences in disease phenotypes and the effectiveness of potential drugs in clinical trials. To help translate research from animal models into novel treatments for ataxia patients, it is essential to validate findings in the relevant affected human cell types, particularly in cerebellar Purkinje cells. The current obstacles might be overcome by exploiting recently developed human induced pluripotent stem cell (iPSC) technology and neuronal differentiation protocols.
Generation of cerebellar neurons using human iPSCs
Major advances in the field of stem cell technology have allowed for the robust reprogramming of human somatic cells into iPSCs10-12. Human ataxia patient-specific iPSCs, which can subsequently be differentiated into specific neuronal subtypes and other affected cell types in cerebellar ataxias, would be an ideal tool for studying the pathogenic mechanisms in human and disease-relevant neurons, and might also be highly valuable for high-throughput drug screening in vitro13,14. Cerebellar Purkinje cells are particularly vulnerable in cerebellar ataxia but difficult to differentiate in vitro, owing to their large size, complex morphology, unique firing properties and extensive period of maturation8,13. However, significant breakthroughs have been achieved recently and are reviewed below.
The generation of cerebellar neurons from human iPSCs aims to recapitulate the complex in vivo molecular events that direct the specification of cerebellar neurons, and circuit formation during human embryogenesis15-17. Early protocols attempted cerebellar differentiation using either mouse or human embryonic stem cells (ESCs)18-21. These approaches sought to mimic the in vivo signals involved in early cerebellar development by delivering inductive signals, including bone morphogenic proteins, mitogens and neurotrophins, in a step-wise fashion throughout the differentiation process. However, the resulting cerebellar cultures only contained a small proportion of Purkinje cells and a relatively high number of cerebellar granule cells18,20.
Muguruma et al. adopted a different approach, which aimed to induce endogenous signals mimicking cerebellar patterning, by adding a combination of only three factors (insulin, fibroblast growth factor 2 (Fgf2) and cyclopamine) to floating aggregates of mouse or human ESCs22,23. Followed by dissociation, fluorescence-activated cell sorting for KIRREL2/NEPH3-positive Purkinje cell progenitors, and subsequent co-culture with mouse primary rhombic lip-derived granule cell precursors, this method resulted in the generation of Purkinje cells at a much higher efficiency than had previously been achieved. The differentiated cells expressed mature PC markers including L7/PCP2 and GRID2, and possessed a characteristic Purkinje cell morphology with an extensively arborized dendritic tree. Moreover, these cells displayed the distinctive firing pattern of Purkinje cells, suggesting that they were functionally mature22,23.
Wang et al. used a similar self-inductive approach to produce Purkinje cells from human iPSCs co-cultured with human foetal cerebellar slices24. Although these cells appeared to display Purkinje cell-specific markers and were electrically active, they did not possess the characteristic morphology of mature Purkinje cells. Moreover, this protocol is likely to be limited in its uptake due to associated ethical issues.
Most recently, the Mugumura group used a very similar approach to their original method to generate Purkinje cells from iPSCs, but with the inclusion of two additional growth factors (brain-derived neurotrophic factor (BDNF) and neurotrophin-3 (NT-3)) in their maturation medium25. After co-culture with mouse primary granule cell precursors for 75 days, the human iPSC-derived neurons possessed extensively branched dendritic trees with a thick stem, characteristic of mature Purkinje cells, and expressed the mature PC-specific markers L7/PCP2 throughout soma and dendrites, and the synaptic receptor GRID2 on dendritic spines. Thus, this protocol not only offers a shorter and more feasible protocol to generate Purkinje cells, but also produces more mature looking PCs that more closely resemble those in vivo.
Despite this significant progress, a number of challenges remain to be addressed before researchers can fully exploit iPSC-based cerebellar models. Dissociated cultures of neurons allow for close monitoring of their individual development throughout differentiation; however, their maturation and survival is limited compared to neurons developing in the embryonic brain, likely due to the lack of appropriate support in in vitro cultures26,27. Moreover, disease phenotypes affecting synapse formation, dendritic retraction and neuronal migration28,29, which may only arise from interactions between cells and their environment, may be lost in these two-dimensional cultures.
A way forward to overcome this limitation might be the use of organoids, self-organizing in vitro structures that include multiple cell types and mimic the in vivo architecture of an organ30. Despite the relative success in generating organoids mimicking development of the cerebral cortex (reviewed in30,31), only one study has thus far reported the generation of cerebellar organoids. Muguruma et al. showed that the addition of Fgf19 and stromal cell-derived factor 1 (Sdf1) during differentiation promoted the self-organisation of human ESCs into polarized three-layer structures reminiscent of the human embryonic cerebellum23. To date, however, no studies have reported the generation of cerebellar organoids from human iPSCs. Moreover, it remains a question whether self-induction alone is sufficient for generating a functionally mature human cerebellum in vitro. While long-term culture is needed for the maturation of multiple cell types that form active neuronal networks in spatially organized manner, the lack of mechanical support, as well as nutrient and gas exchange, cause cell death within organoids and therefore limit culture duration and maturity31. To overcome this, cerebellar organoids could be grown in spinning bioreactors to prolong their viability by improving nutrient and gas exchange32. Alternatively, the self-induction approach could be used to generate progenitors of various cerebellar subtypes, which could subsequently be plated onto biodegradable scaffolds to support their organization into a layered three-dimensional structure30. In such a system, microenvironmental niches surrounding progenitors have been shown to promote terminal differentiation and neuronal circuit formation28.
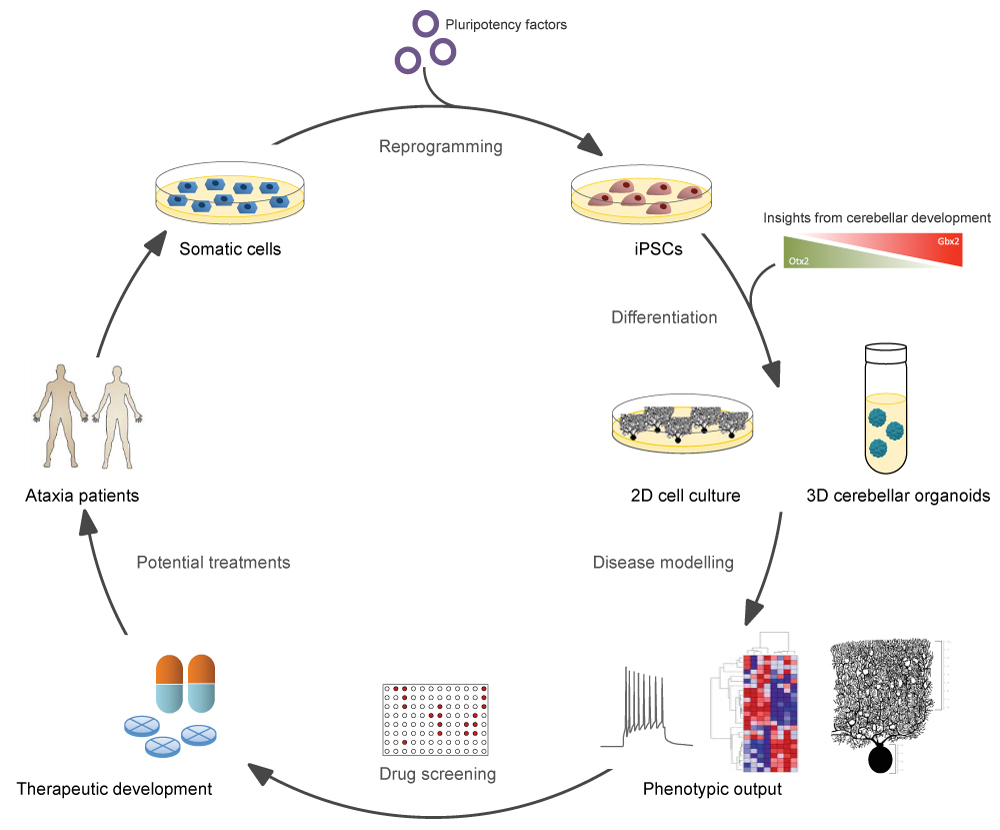
Figure 1:Schematic overview of iPSC-based disease modelling for cerebellar ataxias. Somatic cells from patients are reprogrammed into iPSCs by the introduction of pluripotency factors. These iPSCs can then be differentiated into disease-relevant cell types in either 2D- or 3D-format, following protocols that mimic cerebellar developmental cues in vitro. Cell culture models may then be used as a platform for the investigation of disease phenotypes, as well as for drug screening, with the ultimate goal of delivering effective therapies to patients.
Another limitation of the studies so far is the lack of information on the relative maturity of the generated Purkinje cells and their resemblance to adult human Purkinje cells in vivo. Indeed, transcriptomic analysis of other iPSC-derived neuronal models has revealed that the generated neurons are still in an embryonic state, despite the fact that individual neurons have been stained for mature markers (reviewed in29). Such immature cells may be beneficial for the study of developmental aspects of disease. However, fully mature and functional Purkinje cells will be needed to study the cerebellar degeneration in ataxias due to the late disease onset in patients.
Lastly, the reproducibility of the developed protocols remains a challenge. Although revolutionary in the field, none of the abovementioned studies have been reproduced by other research groups, likely due to their complexity and long differentiation times. Moreover, substantial differences between batches of iPSC differentiation experiments remain a general issue in the field27. To allow for the extensive applications of iPSC-based models, therefore, it is essential to explore ways to shorten the differentiation process, facilitate the maturation of cerebellar neurons and reduce the variability of the protocols. Future studies should focus on detailed electrophysiology, as well as transcriptomic, proteomic and metabolomic analyses of iPSC-derived Purkinje cells. Moreover, it will be vital to compare the characteristics of the generated cerebellar cells to available data from human embryonic and adult cerebellum to stage the iPSC-derived models appropriately.
Disease modelling of cerebellar ataxias
The iPSC-based cerebellar cultures described offer pioneering opportunities to study and model the disease processes underlying cerebellar ataxias in affected human neuronal cell types. Interestingly, mounting evidence from cell and animal models indicates that abnormal Purkinje cell development, including impaired dendritic arborisation, spine development and synaptogenesis, and related functional changes, may contribute to the pathogenesis of ataxias, including neurodegenerative ataxias15. Thus, it will be extremely interesting to investigate whether iPSC-derived human cerebellar neurons display any developmental abnormalities. In addition, these cultures will allow us, for the first time, to unravel the processes that might underlie the cell-specific vulnerability of human cerebellar Purkinje cells.
To date, fewer than 20 studies have reported investigations into cerebellar ataxia, including FRDA, A-T and polyglutamine SCAs, using human iPSCs. The majority of these studies have been performed in neurons without regional specification, as well as in other affected cell types (reviewed in13,14). None of these studies have yet successfully recapitulated the specific cerebellar neuronal dysfunction and degeneration known to characterize these conditions.
In human iPSC-based studies of FRDA, which is caused by an intronic repeat expansion in the FXN gene encoding Frataxin, disease-relevant phenotypes such as reduced Frataxin mRNA and protein levels as well as mitochondrial defects were observed in cardiomyocytes and peripheral sensory neurons, two of the affected cell types in FRDA33-36. In the case of A-T, chromosomal instability and cell cycle checkpoint defects, resulting from mutations in the ATM gene encoding ataxia-telangiectasia mutated (ATM) protein, have been found to reduce the efficiency of the reprogramming of A-T iPSCs37. Although A-T iPSCs can be induced to differentiate into neuronal cells37, it remains to be seen whether they are able to generate Purkinje cells.
A handful of studies have focused on the characterization of iPSCs or iPSC-derived neurons from patients with polyglutamine SCAs, including SCA2, 3, 6 and 725,38-41. To date, only one study has examined the disease phenotype in the context of the affected neuronal cell type. Ishida et al. recently reported the differentiation of iPSCs into Purkinje cells from SCA6 patients that harbour a polyglutamine expansion in the calcium channel Cav2.125. Patient Purkinje cells were more vulnerable to depletion of thyroid hormone triiodothyronine (T3), an important growth hormone for maturation and maintenance of Purkinje cells, as demonstrated by a decrease in cell number and reduction in dendritic arbours in patient cells compared to controls15,42. This study highlights the potential of patient iPSC-derived Purkinje cells to model developmental and disease-relevant ataxia phenotypes. Of note, the SCA6 Purkinje cells only showed abnormal dendritic development upon stress but not under normal culture conditions, suggesting that environmental triggers might be crucial to causing disease phenotypes. It will be interesting to see whether SCA6 phenotypes that have been observed in human post-mortem brain material as well as in SCA6 animal models, including calcium overload, cytoplasmic aggregation of expanded Cav2.1 protein, nuclear localization of the cleaved Cav2.1 C-terminus and the resulting perturbation of gene expression5,43, can be recapitulated in the iPSC-derived SCA6 model.
Notably, the observed developmental phenotype in the SCA6 Purkinje cells could be rescued using two compounds, thyroid releasing hormone and riluzole, demonstrating the potential of patient-specific iPSC-derived cerebellar neurons to be used for drug screening and personalized therapy.
Conclusion
Recent breakthroughs in generating human iPSC-derived cerebellar neurons offer unprecedented opportunities to study the disease mechanisms underlying cerebellar ataxias. This exciting methodology will be particularly powerful when combined with other cutting-edge technologies including genome engineering, single-cell sequencing and high-resolution microscopy. Together, iPSC-derived cerebellar models are poised to advance our understanding of the disease mechanisms underlying the cerebellar ataxias, thereby enabling the development of more effective treatment options for patients in the future.
Acknowledgement
Research in the Becker laboratory is funded by the Royal Society, the Wellcome Trust, the European Commission (Marie Sklodowska-Curie Actions), the Rosetrees Trust, BrAshA-T and the John Fell OUP Research Fund.
Conflict of Interest Statement
We declare no conflict of interests.
References
- Rossi M, Perez Lloret S, Doldan L, et al. Autosomal dominant cerebellar ataxias: a systematic review of clinical features. Eur J Neurol. 2014; 21(4): 607-615.
- Bürk K. Clinical Scales of Cerebellar Ataxias. In: Manto M, Gruol DL, Schmahmann JD, et al, editors. Handbook of the Cerebellum and Cerebellar Disorders. Dordrecht: Springer Netherlands; 2013. pp. 1783-1798.
- Sun YM, Lu C, Wu ZY. Spinocerebellar ataxia: relationship between phenotype and genotype a review. Clin Genet. 2016; 90(4): 305-314.
- Sandford E, Burmeister M. Genes and genetic testing in hereditary ataxias. Genes. 2014; 5(3): 586-603.
- Matilla Duenas A, Ashizawa T, Brice A, et al. Consensus paper pathological mechanisms underlying neurodegeneration in spinocerebellar ataxias. Cerebellum. 2014; 13(2): 269-302.
- Meera P, Pulst SM, Otis TS. Cellular and circuit mechanisms underlying spinocerebellar ataxias. J Physiol. 2016; 594(16): 4653-4660.
- Bushart DD, Murphy GG, Shakkottai VG. Precision medicine in spinocerebellar ataxias treatment based on common mechanisms of disease. Ann Transl Med. 2016; 4(2): 25.
- Hekman KE, Gomez CM. The autosomal dominant spinocerebellar ataxias: emerging mechanistic themes suggest pervasive Purkinje cell vulnerability. J Neurol Neurosurg Psychiatry. 2015; 86(5): 554-561.
- Cendelin J. From mice to men lessons from mutant ataxic mice. Cerebellum Ataxias. 2014; 1: 4.
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006; 126(4): 663-676.
- Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007; 131(5): 861-872.
- Yu J, Vodyanik MA, Smuga Otto K, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007; 318(5858): 1917-19120.
- Watson LM, Wong MMK, Becker EBE. Induced pluripotent stem cell technology for modelling and therapy of cerebellar ataxia. Open Biol. 2015; 5(7): 150056.
- Lukovic D, Moreno Manzano V, Rodriguez Jimenez FJ, et al. hiPSC Disease Modeling of Rare Hereditary Cerebellar Ataxias. Neuroscientist. 2016; 1073858416672652.
- Leto K, Arancillo M, Becker EBE, et al. Consensus Paper: Cerebellar Development. Cerebellum. 2016; 15(6): 789-828.
- Butts T, Green MJ, Wingate RJ. Development of the cerebellum: simple steps to make a 'little brain'. Development. 2014; 141(21): 4031-4041.
- Andreu A, Crespo Enriquez I, Echevarria D. Molecular events directing the patterning and specification of the cerebellum. Eur J Anat. 2014; 18(4): 245-252.
- Erceg S, Ronaghi M, Zipancic I, et al. Efficient differentiation of human embryonic stem cells into functional cerebellar-like cells. Stem Cells Dev. 2010; 19(11): 1745-1756.
- Erceg S, Lukovic D, Moreno Manzano V, et al. Derivation of cerebellar neurons from human pluripotent stem cells. Curr Protoc Stem Cell Biol. 2012; Chapter 1: Unit 1H.5.
- Salero E, Hatten ME. Differentiation of ES cells into cerebellar neurons. Proc Natl Acad Sci U S A. 2007; 104(8): 2997-3002.
- Su HL, Muguruma K, Matsuo Takasaki M, et al. Generation of cerebellar neuron precursors from embryonic stem cells. Dev Biol. 2006; 290(2): 287-296.
- Muguruma K, Nishiyama A, Ono Y, et al. Ontogeny-recapitulating generation and tissue integration of ES cell-derived Purkinje cells. Nat Neurosci. 2010; 13(10): 1171-1180.
- Muguruma K, Nishiyama A, Kawakami H, et al. Self-organization of polarized cerebellar tissue in 3D culture of human pluripotent stem cells. Cell Rep. 2015; 10(4): 537-550.
- Wang S, Wang B, Pan N, et al. Differentiation of human induced pluripotent stem cells to mature functional Purkinje neurons. Sci Rep. 2015; 5: 9232.
- Ishida Y, Kawakami H, Kitajima H, et al. Vulnerability of Purkinje Cells Generated from Spinocerebellar Ataxia Type 6 Patient-Derived iPSCs. Cell Rep. 2016; 17(6): 1482-1490.
- Mariani J, Coppola G, Zhang P, et al. FOXG1-Dependent Dysregulation of GABA/Glutamate Neuron Differentiation in Autism Spectrum Disorders. Cell. 2015; 162(2): 375-390.
- Quadrato G, Nguyen T, Macosko EZ, et al. Cell diversity and network dynamics in photosensitive human brain organoids. Nature. 2017; 545(7652): 48-53.
- Birey F, Andersen J, Makinson CD, et al. Assembly of functionally integrated human forebrain spheroids. Nature. 2017; 545(7652): 54-59.
- Ardhanareeswaran K, Mariani J, Coppola G, et al. Human induced pluripotent stem cells for modelling neurodevelopmental disorders. Nat Rev Neurol. 2017; 13(5): 265-278.
- Dutta D, Heo I, Clevers H. Disease Modeling in Stem Cell-Derived 3D Organoid Systems. Trends Mol Med. 2017; 23(5): 393-410.
- Kelava I, Lancaster MA. Dishing out mini-brains: Current progress and future prospects in brain organoid research. Dev Biol. 2016; 420(2): 199-209.
- Qian X, Nguyen HN, Song MM, et al. Brain-Region-Specific Organoids Using Mini-bioreactors for Modeling ZIKV Exposure. Cell. 2016; 165(5): 1238-1254.
- Eigentler A, Boesch S, Schneider R, et al. Induced pluripotent stem cells from friedreich ataxia patients fail to upregulate frataxin during in vitro differentiation to peripheral sensory neurons. Stem Cells Dev. 2013; 22(24): 3271-3282.
- Hick A, Wattenhofer Donze M, Chintawar S, et al. Neurons and cardiomyocytes derived from induced pluripotent stem cells as a model for mitochondrial defects in Friedreich's ataxia. Dis Model Mech. 2013; 6(3): 608-621.
- Liu J, Verma PJ, Evans Galea MV, et al. Generation of induced pluripotent stem cell lines from Friedreich ataxia patients. Stem Cell Rev. 2011; 7(3): 703-713.
- Du J, Campau E, Soragni E, et al. Role of mismatch repair enzymes in GAA.TTC triplet-repeat expansion in Friedreich ataxia induced pluripotent stem cells. J Biol Chem. 2012; 287(35): 29861-29872.
- Nayler S, Gatei M, Kozlov S, et al. Induced pluripotent stem cells from ataxia-telangiectasia recapitulate the cellular phenotype. Stem Cells Transl Med. 2012; 1(7): 523-535.
- Morino H, Matsuda Y, Muguruma K, et al. A mutation in the low voltage-gated calcium channel CACNA1G alters the physiological properties of the channel, causing spinocerebellar ataxia. Mol Brain. 2015; 8: 89.
- Xia G, Santostefano K, Hamazaki T, et al. Generation of human-induced pluripotent stem cells to model spinocerebellar ataxia type 2 in vitro. J Mol Neurosci. 2013; 51(2): 237-248.
- Koch P, Breuer P, Peitz M, et al. Excitation-induced ataxin-3 aggregation in neurons from patients with Machado-Joseph disease. Nature. 2011; 480(7378): 543-546.
- Luo Y, Fan Y, Zhou B, et al. Generation of induced pluripotent stem cells from skin fibroblasts of a patient with olivopontocerebellar atrophy. Tohoku J Exp Med. 2012; 226(2): 151-159.
- Heuer H, Mason CA. Thyroid hormone induces cerebellar Purkinje cell dendritic development via the thyroid hormone receptor alpha1. J Neurosci. 2003; 23(33): 10604-10612.
- Taroni F, Chiapparini L, Mariotti C. Autosomal Dominant Spinocerebellar Ataxias and Episodic Ataxias. In: Manto M, Gruol DL, Schmahmann JD, et al, editors. Handbook of the Cerebellum and Cerebellar Disorders. Dordrecht: Springer Netherlands; 2013. pp. 2193-2267.