
The two sides of Faim2 -- modulation of cell death and regeneration
Björn Falkenburger and Arno Reich
Most neurologic diseases are associated with neuronal cell death, and the surviving neurons consequently initiate regenerative processes. Activation of Fas/CD95, a so-called death receptor, can induce both apoptosis and alternative, non-apoptotic intracellular signaling1. Consequently, modulators of the Fas/CD95 signaling pathways have gained importance. We have investigated the role of one such mudulator, Fas inhibitory molecule 2 (Faim2), in neurologic diseases. We generated a Faim2 deficient mouse line and applyed models of transient cerebral ischemia, bacterial meningitis, and Parkinson disease2-4. In brief, we found increased neuronal cell death in the acute phase of all disease models in Faim2 deficient mice in comparison to the Faim2 wildtype (WT). In addition, signs of increased regeneration in Faim2 deficient mice point towards an involvement of Faim2 and Fas/CD95 in regenerative processes. Finally, we found disease stage-dependent regulation of Faim2 expression, potentially enabling the switch between apoptotic and alternative Fas/CD95 signaling.
Thus, Faim2 appears to be an interesting protein for two reasons. First, it could be a target protein for neuroprotective strategies in the above mentioned diseases. Second, it may help us to understand the cellular signaling pathways involved in neuronal cell death and in regeneration. In spite of its name, Faim2 is not well known. We therefore start with nomenclature issues before describing the neurobiology of Faim2 in more detail.
What do we know about Faim2?
Faim2 is a member of two different protein families. The first family is defined functionally as Fas inhibitory molecules and comprises 3 members (Faim1, Faim2, Faim3) that are structurally unrelated and show different expression patterns throughout the organism5,1. In addition, Faim2 is a member of a structurally defined family of proteins related to apoptosis named transmembrane BAX inhibitor-1 motif-containing (TMBIM) proteins, or life guard (Lfg) proteins. All family members (TMBIM1/Lfg3, TMBIM1b/Lfg5, TMBIM2/Lfg2, TMBIM3/Lfg1, TMBIM4/Lfg4) are highly conserved throughout evolution. Faim2 is TMBIM2. Another synonym for Faim2 is neuronal membrane protein 35 (NMP35) due to its molecular weight of 35 kD. TMBIM3 is also called Glutamate Receptor, Ionotropic, N-Methyl D-Aspartate-Associated Protein 1 (GRINA1). Among the TMBIM proteins, Faim2 and GRINA1 have been studied most. All TMBIM proteins have been associated with various intracellular processes, including apoptosis, calcium homeostasis, unfolded protein response, autophagy and lysosomal function, which is reflected in the confusing nomenclature6. Their precise functions have not been fully resolved.
Faim2 is primarily expressed in neurons7,8. The constitutive expression of Faim2 in cultured cerebellar granule neurons prevents apoptotic cell death induced Fas ligand9. Protective effects of Faim2 in cultured neurons have also been described for Purkinje cells and cortical neurons10,7. In addition to these neuronal effects, Faim2 single nucleotide polymorphisms (SNPs) have been linked to an increased susceptibility for obesity, especially in childhood, in a genome wide association study (GWAS)11. For certain subpopulations, i.e. typ-2 diabetic patients, Faim2 SNPs have been linked a to higher risk of myocardial infarction12. In accordance with its inhibition of apoptosis, Faim2 was found associated with the biology of certain types of tumors, including breast cancer13, neuroblastoma14 and small-cell lung cancer15, possibly with therapeutic implications16. This review focuses, however, on the role of Faim2 in non neoplastic neurological diseases.
Faim2 is protective in animal models of neurologic diseases
In an animal model of transient and focal cerebral ischemia, ischemia was induced by occlusion of the middle cerebral artery for 30 minutes followed by 72 hours of reperfusion. Cell death was associated with caspase-8 and caspase-3 activation, as expected for Fas dependent apoptosis. Faim2 deficient mice showed larger infarcts than Faim2 WT mice and a higher number of apoptotic neurons. This effect was rescued by lentiviral overexpression of Faim2 in the null mutant, in accordance with an inhibition of Fas dependent apoptosis by Faim2. Interestingly, lentiviral overexpression of Faim2 also reduced infarct size in WT mice, and Faim2 expression was transiently downregulated 18 h after artery occlusion2.
In an animal model of bacterial meningitis, Faim2 deficient mice showed a higher number of apoptotic neurons in the hippocampus than WT littermates 24 hours after infection with Streptococcus pneumoniae. Animals were treated with antibiotics twice daily starting 18 hours after infection. Neuronal degeneration in this model occurs primarily in the hippocampus and is associated with a learning deficit after recovery. Interestingly, Faim2 expression was reduced to 50% 20 hours after infection, echoing the findings obtained in the ischemia model. As an indication of alternative Fas signaling we found increased ERK phosphorylation and a higher number of new (BrdU positive) neurons in the hippocampus of Faim2 deficient mice more than 8 weeks after infection. Accordingly, Faim2 deficient mice performed better than wildtype mice in a learning paradigm 8 weeks after bacterial meningitis. The relevance of these findings for human patients was supported by the fact that we found more Faim2 positive neurons in the hippocampus of patients with bacterial meningitis than in controls3.
Finally, in the classic animal model of Parkinson disease, Faim2 deficient mice displayed a more pronounced loss of TH positive dopaminergic neurons in the substantia nigra than WT. Mice were analyzed 14 days after injecting the dopaminergic cell specific neurotoxin MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine), applied in 5 injections of 30 mg/kg spaced 24 hours apart. 14 days after MPTP we also found a more pronounced loss of dopaminergic axon terminals in the striatum of Faim2 deficient mice. Dopaminergic axon terminals recover by sprouting between 14 and 90 days after the last MPTP injection. This regeneration was more pronounced in Faim2 deficient mice, consistent with a potential role of Fas/CD95 in neuron regeneration. Faim2 expression was decreased by MPTP administration, again similar as in cerebral ischemia and bacterial meningitis4.
Towards a Faim2 based neuroprotective therapy
Taken together, we saw evidence of neuroprotection by Faim2 in three animal models of neurologic diseases. Common to these models is the occurrence of delayed and programmed cell death (see Figure 1). In our model of transient cerebral ischemia only the cells in the core of the infarct degenerate due to immediate glucose and oxygen deprivation. Most of the cells in the penumbra, however, degenerate through other mechanisms, including excitotoxicity, reactive oxygen species, and inflammation17. Similarly, cell death following bacterial meningitis is not primarily mediated by bacterial invasion, but by the inflammatory response that follows infection3. Finally, neurons in Parkinson disease and in the applied subacute MPTP model die by an apoptotic mechanism with contributions from excitotoxicity, oxidative stress and inflammation18,19. This delayed and programmed cell death can potentially be modulated by Faim2.
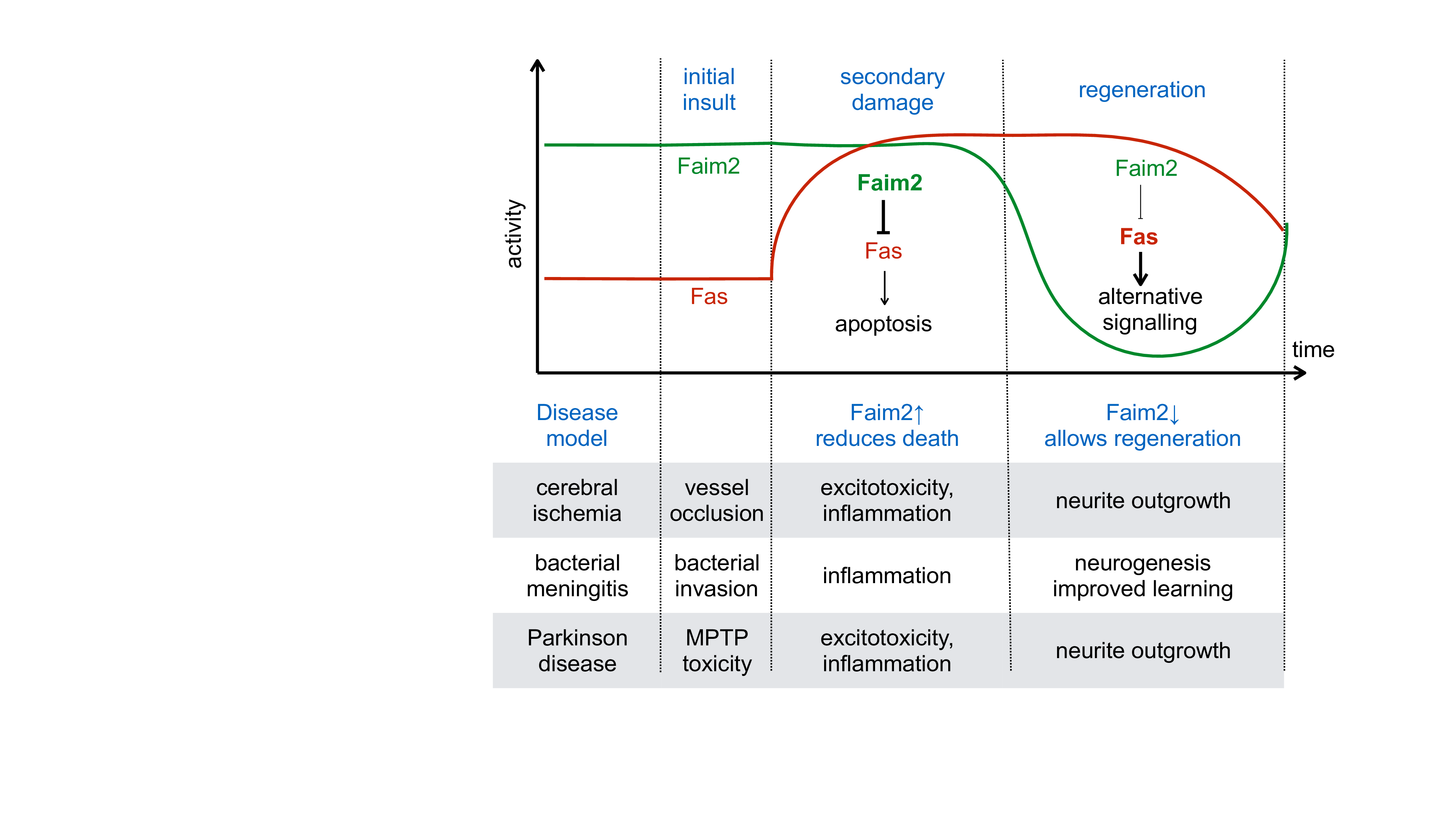
Figure 1: Schematic representation of the regulation of Fas and Faim2 during the different phases of the neurologic diseases studied. Faim2 is constitutitvely expressed, inhibiting Fas dependent apoptotic cell death. After the acute insult, delayed and programmed cell death occurrs by excitotoxicity, oxidative stress and inflammation. In this phase, constitutive Faim2 can prevent Fas induced cell death, and increasing Faim2 can further reduce cell death. In the last phase, however, down regulation of Faim2 may allow alternative Fas signalling that underlies regenerative processes such as proliferation and differentiation of neuronal precursors and neurite outgrowth.
Two findings indicate that there a realistic therapeutic potential for neuroprotection by Faim2. First, in the study of transient cerebral ischemia, lentiviral overexpression of Faim2 decreased the infarct size in wildype mice. This indicates that an increase in Faim2 may have protective effects even when Faim2 is constitutively expressed. Moreover, we have found in all three animal models a down regulation of Faim2. Therefore, increasing Faim2 or preventing Faim2 downregulation in early disease processes may attenuate cell death.
What do we need in order to develop this therapeutic option further? First, we need to know what regulates Faim2 expressionsp that we can increase Faim2 expression in the disease state. Faim2 expression in cultured cells was regulated by the PI3-kinase / Akt pathway9. Since this pathway is downstream of several well known growth factor receptors, it seems a plausible next step to test the therapeutic potential of agents stimulating these pathways. One of these pathways is erythropoietin, for which beneficial effects have been observed in animal models of cerebral ischemia20,21 and Parkinson disease22,23.
Second, it will be beneficial to learn by which molecular mechanism Faim2 prevents cell death. Faim2 has been shown to physically interact with Fas and prevent downstream activation of caspases7,8,24. Still, it is unclear whether the interaction of Faim2 with Fas prevents Fas trimerization, ligand binding, trafficking, or formation of the "death inducing signaling complex" with FADD and the initiator caspase 8. Moreover, some recent evidence indicates that Faim2 modulates cytosolic calcium as do other members of the TMBIM family25,26. Interestingly, regulation of calcium is also important for cell death in models of cerebral ischemia and Parkinson disease27-29.
Third, we have found evidence that deficiency of Faim2 or its downregulation can be associated with beneficial effects in the later phases of disease models. We found more neurogenesis (BrdU positive neurons) in the hippocampus 8 weeks after pneumococcal infection, improved learning after meningitis, and more pronounced sprouting 90 days after MPTP intoxication3,4. These findings are consistent with the now established role of Fas/CD95 for regenerative processes, including proliferation and differentiation of neuronal precursors and neurite outgrowth30,24,31. It is tempting to speculate that facilitating these regenerative effects of Fas/CD95 signaling is the "purpose" of the Faim2 downregulation observed in disease states. In order to further develop a therapeutic strategy based on Faim2, it will therefore be crucial to determine when in the course of a disease Faim2 upregulation will be beneficial and when Faim2 inhibition would be more appropriate. One option to investigate this hypothesis will be to cross mice carrying the floxed Faim allele1 with mice in which Cre recombinase can be induced by tamoxifen administration. It is likely though, that the time windows for beneficial and detrimental Faim2 activity differ between disease models and between mice and man.
Conclusion
Faim2 is constitutively expressed in neurons and inhibits Fas/CD95 signaling. It is therefore in an excellent position to prevent detrimental Fas/CD95 signaling and apoptotic cell death in response to a sublethal insult, and to allow Fas/CD95 signaling stimulating regenerative processes. However, it is exactly this dual function that makes it difficult to use Faim2 for neuroprotection. It is possible that further strategies tested for neuroprotection can similarly elicit beneficial and detrimental effects. Whether neuroprotection prevails may then depend on timing as outlined for Faim2, or may relate do dosing in other cases. In order to overcome this problem we need to understand for each therapeutic strategy the signaling pathway that mediates the beneficial and detrimental effects. We will then be able to develop biomarkers for both branches of signaling and determine in patients optimal dosing and timing for these therapeutic strategies.
References
- Reich A, Spering C, Schulz JB. Death receptor Fas (CD95) signaling in the central nervous system tuning neuroplasticity. Trends in Neurosciences. 2008 Sep; 31(9): 478–86.
- Reich A, Spering C, Gertz K, et al. Fas/CD95 regulatory protein Faim2 is neuroprotective after transient brain ischemia. Journal of Neuroscience. 2011 Jan 5; 31(1): 225–33.
- Tauber SC, Harms K, Falkenburger BH, et al. Modulation of Hippocampal Neuroplasticity by Fas/CD95 Regulatory Protein 2 (Faim2) in the Course of Bacterial Meningitis. J Neuropathol Exp Neurol. 2014 Jan; 73(1): 2–13.
- Komnig D, Schulz JB, Reich A, Falkenburger BH. Mice lacking Faim2 show increased cell death in the MPTP mouse model of Parkinson disease. Journal of neurochemistry. 2016 Dec;139(5):848–57.
- Planells-Ferrer L, Urresti J, Coccia E, et al. Fas apoptosis inhibitory molecules: more than death-receptor antagonists in the nervous system. Journal of neurochemistry. 2016 Oct; 139(1): 11–21.
- Rojas-Rivera D, Hetz C. TMBIM protein family ancestral regulators of cell death. Oncogene. 2015 Jan 15; 34(3): 269–80.
- Fernández M, Segura MF, Solé C, et al. Lifeguard/neuronal membrane protein 35 regulates Fas ligand-mediated apoptosis in neurons via microdomain recruitment. Journal of neurochemistry. 2007 Oct; 103(1): 190–203.
- Schweitzer B, Taylor V, Welcher AA, et al. Neural membrane protein 35 (NMP35) a novel member of a gene family which is highly expressed in the adult nervous system. Mol Cell Neurosci. 1998 Aug; 11(5-6): 260–73.
- Beier CP, Wischhusen J, Gleichmann M, et al. FasL (CD95L/APO-1L) resistance of neurons mediated by phosphatidylinositol 3-kinase-Akt/protein kinase B-dependent expression of lifeguard/neuronal membrane protein 35. Journal of Neuroscience. 2005 Jul 20; 25(29): 6765–74.
- Hurtado de Mendoza T, Perez-Garcia CG, Kroll TT, et al. Antiapoptotic protein Lifeguard is required for survival and maintenance of Purkinje and granular cells. Proceedings of the National Academy of Sciences. 2011 Oct 11; 108(41): 17189–94.
- Felix JF, Bradfield JP, Monnereau C, et al. Genome-wide association analysis identifies three new susceptibility loci for childhood body mass index. Human Molecular Genetics. 2016 Jan 15; 25(2): 389–403.
- Corella D, Sorlí JV, González JI, et al. Novel association of the obesity risk-allele near Fas Apoptotic Inhibitory Molecule 2 (FAIM2) gene with heart rate and study of its effects on myocardial infarction in diabetic participants of the PREDIMED trial. Cardiovasc Diabetol. 2014 Jan 6; 13: 5.
- Bucan et al., Apotosis 2010; Bucan et al, Cell Mol Biol Lett 2010 Bucan et al, Oncol Left 2011.
- Planells-Ferrer L, Urresti J, Soriano A, et al. MYCN repression of Lifeguard/FAIM2 enhances neuroblastoma aggressiveness. Cell Death Dis. 2014 Sep 4; 5: e1401.
- Kang HC, Kim JI, Chang HK, et al. FAIM2, as a novel diagnostic maker and a potential therapeutic target for small-cell lung cancer and atypical carcinoid. Sci Rep. 2016 Sep 28; 6: 34022.
- Radin D, Lippa A, Patel P, et al. Lifeguard inhibition of Fas-mediated apoptosis A possible mechanism for explaining the cisplatin resistance of triple-negative breast cancer cells. Biomed Pharmacother. 2016 Feb; 77: 161–6.
- Eberhardt O, Coelln RV, Kugler S, et al. Protection by synergistic effects of adenovirus-mediated X-chromosome-linked inhibitor of apoptosis and glial cell line-derived neurotrophic factor gene transfer in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson's disease. Journal of Neuroscience. 2000 Dec 15; 20(24): 9126–34.
- Hald A, Lotharius J. Oxidative stress and inflammation in Parkinson's disease: is there a causal link. Exp Neurol. 2005 Jun; 193(2): 279–90.
- Byts N, Sirén AL. Erythropoietin a multimodal neuroprotective agent. Exp Transl Stroke Med. 2009 Oct 21; 1: 4.
- Minnerup J, Heidrich J, Rogalewski A, et al. The Efficacy of Erythropoietin and Its Analogues in Animal Stroke Models A Meta-Analysis. Stroke. 2009 Aug 24; 40(9): 3113–20.
- Huang CK, Chang YT, Amstislavskaya TG, et al. Synergistic effects of ceftriaxone and erythropoietin on neuronal and behavioral deficits in an MPTP-induced animal model of Parkinson's disease dementia. Behavioural Brain Research. 2015 Nov 1; 294: 198–207.
- Schweitzer B, Suter U, Taylor V. Neural membrane protein 35/ Lifeguard is localized at postsynaptic sites and in dendrites. Molecular Brain Research. 2002 Oct; 107(1): 47–56.
- Somia NV. LFG an anti-apoptotic gene that provides protection from Fas-mediated cell death. Proc Natl Acad Sci USA. 1999 Oct 26; 96(22): 12667–72.
- Schweitzer B, Suter U, Taylor V. Neural membrane protein 35/Lifeguard is localized at postsynaptic sites and in dendrites. Molecular Brain Research. 2002 Oct; 107(1): 47–56.
- Urresti J, Ruiz-Meana M, Coccia E, et al. Lifeguard Inhibits Fas Ligand-mediated Endoplasmic Reticulum-Calcium Release Mandatory for Apoptosis in Type II Apoptotic Cells. J Biol Chem. 2016 Jan 15; 291(3): 1221–34.
- Sulzer D, Surmeier DJ. Neuronal vulnerability pathogenesis and Parkinson's disease. Mov Disord. 2013 Jun; 28(6): 715–24.
- Dragicevic E, Schiemann J, Liss B. Dopamine midbrain neurons in health and Parkinson's disease emerging roles of voltage gated calcium channels and ATP sensitive potassium channels. Neuroscience. 2015 Jan 22; 284: 798–814.
- Bodalia A, Li H, Jackson MF. Loss of endoplasmic reticulum Ca2+ homeostasis: contribution to neuronal cell death during cerebral ischemia. Acta Pharmacol Sin. 2013 Jan; 34(1): 49–59.
- Desbarats J, Birge RB, Mimouni-Rongy M, et al. Fas engagement induces neurite growth through ERK activation and p35 upregulation. Nature Cell Biology. 2003 Jan 20; 5(2): 118–25.
- Zuliani C, Kleber S, Klussmann S, et al. Control of neuronal branching by the death receptor CD95 (Fas/Apo-1). Cell Death Differ. 2006 Jan; 13(1): 31–40.
Abbreviations:
| Akt | protein kinase B |
| BrdU | Bromodeoxyuridine |
| Cre | causes recombination of two LoxP (locus of X-over P1) sites |
| ERK | Extracellular Signal-regulated Kinase |
| FADD | Fas associated protein with death domain |
| Faim2 | Fas inhibitory molecule 2 |
| floxed | flanked by LoxP (locus of X-over P1) sites |
| GWAS | genome wide association study |
| Lfg | life guard |
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| PI3-kinase | phosphatidylinositol 3-kinase |
| SNP | single-nucleotide polymorphism |
| TMBIM | transmembrane BAX inhibitor-1 motif-containing |
| WT | wild type |