
Anti-Iglon5 Syndrome: What We Know So Far? A Non-Systematic Review
Caroline Figueiredo da Silva1*, Gustavo Figueiredo da Silva1, Washigton Luiz Gomes de Medeiros Junior1, Marcus Vinícius Magno Gonçalves2
1Medical student - Department of Medicine, University of the Region of Joinville (UNIVILLE), Brazil
2Medical Doctor, PhD and Professor of Neurology, University of the Region of Joinville (UNIVILLE), Brazil
Abstract
The objective of this review is to provide an overview of the current knowledge of Anti-IgLON5 syndrome. The IgLON proteins are a family of cell adhesion molecules and the presence of antibodies against IgLON5 is crucial for the Anti-IgLON5 Syndrome diagnosis. This syndrome has an expanded clinical spectrum that involves prominent sleep disorder, progressive bulbar dysfunction, gait instability with abnormal eye movements reminiscent, and cognitive deterioration sometimes associated with chorea. The main neuropathological finding is the neuronal loss with hyperphosphorylated tau (p-Tau) protein accumulation at the hypothalamus, brainstem tegmentum, hippocampus, periaqueductal gray matter, medulla oblongata, and upper cervical cord. The exact pathogenesis is still unclear and involves a neurodegenerative process and autoimmune response. Early diagnosis is important to avoid unnecessary tests and prevent complications. Important resources for diagnosis are the antibody testing of serum and CSF for IgLON5-IgG. The Anti-IgLON5 syndrome mortality is high and new studies published described a good response to immune therapy. However, the response to immune therapy depends on some clinical and analytical characteristics. In addition, future studies are needed to thoroughly analyze the aspects of pathogenesis and treatment of this important pathological syndrome.
Abbreviations
CNS - Central Nervous System
CSF - Cerebrospinal Fluid
IgG - Immunoglobulin G
MRI – Magnetic Resonance Imaging
PSP-like - Progressive Supranuclear Palsy
p-Tau - Hyperphosphorylated Tau Protein
REM - Rapid Eye Movement
Introduction
The first report of Anti-IgLON5 syndrome was in 2014, when antibodies against the cell surface protein IgLON5 were described associated with a complex neurological syndrome of sleep disturbances and movement disorders. Anti-IgLON5 antibodies have a prevalence of 12 in 150,000 patients per year. However, considering the unreported patients, the prevalence can be much higher1. Since 2014, new researches showed an expanded clinical spectrum that involves sleep dysfunction, bulbar dysfunction, chorea, and progressive supranuclear palsy-like symptoms whereas dysautonomia and cognitive decline are presents in more advanced stages2. The objective of this paper is to review the main findings about Anti-IgLON5 syndrome.
Methods
This study is a non-systematic review based on articles published since 2014 in Medline accessed at Pubmed. The discussion divided into points to give an overview of the pathophysiology, clinical manifestations, diagnoses, and treatment of the Anti- IgLON5 syndrome.
Discussion
Understanding Anti-IgLON5 syndrome: The pathophysiology
The IgLON proteins are a family of cell adhesions molecules, containing five different proteins (IgLON 1-5), that belongs to the immunoglobulin domain superfamily. Until today, the pathophysiological function of IgLON5 is unknown3,4. It has been suggested that IgLONs play a role in the evolution of brain anatomy and complex maturation, influencing cellular migration and brain-blood barrier integrity5,6.
Antibodies against IgLON5 are crucial for diagnosis and strongly associated with the HLA-DRB1*1001 and HLA-DQB1*0501 alleles. These antibodies are present in serum (100%) and CSF (94.9%). In addition, Anti-IgLON5 syndrome displays all four isotypes of Immunoglobulin G (IgG), with predominance of IgG4 antibodies. The IgG4 represent a mean of 58% of the total IgG reactive and have a higher density in serum6,7.
An important study conducted by Sabater and colleagues8 in 2016 analyzed the human antibodies associated with the anti-IgLON5 syndrome and their immunological characteristics. In this study, samples from 15 patients with the syndrome were analyzed. The predominant subclass of IgLON5 antibodies was IgG4, but all samples showed a variable amount of IgG1 (33% on average). IgLON5 antibodies recognize immunoglobulin-like domain 2 of the antigen, and the reactivity is not dependent on glycosylation. Besides, the effects observed in hippocampal neuronal cultures indicate irreversible internalization mediated by surface IgLON5 antibodies. Specific IgG1 mediated these effects and was associated with the disease pathogenesis8.
The main neuropathological finding in Anti-IgLON5 disease is the neuronal loss with hyperphosphorylated tau (p-Tau) protein accumulation (3-repeat and 4-repeat isoform). Hypothalamus and brainstem tegmentum were the sites of p-Tau protein greatest aggregation, still with a rostrocaudal gradient involving the hippocampus, periaqueductal gray matter, medulla oblongata, and upper cervical cord. No inflammatory infiltrates or concomitant abnormal protein deposits of beta-amyloid or alpha-synuclein were detected9,10. In contrast, substantia nigra, basal ganglia, cortex, thalamus, and cerebellum are preserved or slightly affected, showing a different anatomical brain distribution than primary tauopathies (e.g., corticobasal syndrome, progressive supranuclear palsy)11.
An important study conducted by Gelpi et al.9 suggested research criteria for the neuropathological Anti-IgLON5 disease diagnosis. The criteria involve neuropathological findings associated with clinical and immunological aspects and classify as “definitive”, “probable” or “possible” diagnosis. The diagnosis is 1) "definitive" when the neuropathological pattern is present with antibodies detection, regardless of CSF or serum; 2) "probable" when neuropathological findings are present, along with clinical history or alleles confirmation (HLA-DRB1*1001 and HLA-DQB1*0501), even with unknown antibody status; and 3) "possible" when neuropathological findings are present without immunological or clinical information9.
The exact pathogenesis of this disorder is still unclear, and a primary neurodegenerative process can trigger the autoimmune response. According to Gaig et al7, the IgLON5 antibody interference in the internal cytoskeletal network can generates neuronal dysfunction, however it is a hypothesis7.
Clinical manifestations
The onset of Anti-IgLON5 disease is insidious and may be between 45 to 75 years12. Moreover, the onset involves progressive sleep and brainstem disorders. Whereas neuropsychiatric disorders, dysautonomia, and hyperexcitability (myoclonus, cramps, and exaggerated startle) are most commonly described in late stages12,13. Most often, the sleep disorder involves, during non-REM (Rapid Eye Movement) and REM, sleep breathing changes and parasomnias while, among the brainstem disorders, gait abnormalities, dysphagia, dysarthria, and oculomotor problems9,12. Furthermore, there are described cognitive impairment, psychiatric symptoms, myoclonus, cramps, and exaggerated startle as hyperexcitability symptoms12.
Thus, four clinical presentations were characterized according to predominant symptoms: a sleep disorder with parasomnia and sleep breathing difficulty, a bulbar syndrome including dysphagia, sialorrhea, stridor, or acute respiratory insufficiency, a syndrome resembling progressive supranuclear palsy (PSP-like) (vertical and horizontal supranuclear gaze palsies) and cognitive decline with or without chorea7 (figure 1).
Despite the evidence that Anti-IgLON5 disease is related to the central nervous system, clinical descriptions suggest that there is an important role of anti-IgLON5 antibodies on peripheral nerves. Thus, peripheral involvement with neuromyotonic discharges, muscle weakness and wasting may be related to Anti-IgLON5 disease also1.
Key points for diagnosis
Based on those symptoms, adults with unexplained sleep disorder, brainstem disorders, dysautonomia, hyperexcitability, and neuropsychiatric disorders should be screened for IgLON5 disease. The early diagnosis is important to avoid unnecessary tests, prevent complications, as acute respiratory failure, and irreversible neuronal damage, and have the potential to improve outcome with the correct treatment7,14.
For screening, antibody testing of serum and CSF for IgLON5-IgG is crucial4. Besides, HLA-DRB1*1001 and HLA-DQB1*0501 are strongly associated to with the presence of anti-IgLON5 antibodies. In a review of reported cases, Nissen MS and colleague 20196 demonstrated that antibodies, in serum and CSF, are positive in 95 and 100% of cases, respectively. Moreover, was demonstrated that brain FDG-PET CT, abnormal in 50% of cases, could be more sensitive than Magnetic Resonance Imaging (MRI). In addition, abnormal polysomnography was found in 95% of cases6.
Additional paraclinical investigations, some findings are infrequent and nonspecific as subacute presentation, autoimmune disease history, electromyography, an inflammatory CSF, or inflammatory-appearing brain imaging. Other tests may be important according to the clinical presentation4,13.
Since the symptoms are nonspecific and wide, it is important to perform differential diagnosis with other diseases. Among the diseases, progressive supranuclear palsy, others tauopathy, Central Nervous System (CNS) Whipple disease, and multiple system atrophy are some examples4,15.
Treatment Resources
Anti-IgLON5 syndrome is a controversial disease where the answer to the pathological mechanism is uncertain. This syndrome can be considered in two ways: 1) as a degenerative disease with a secondary inflammatory response against IgLON5 or 2) as an autoimmune disorder, in which the neuronal accumulation of tau mediated by antibodies results in a functional disorder followed by neurodegeneration16. The Anti-IgLON5 syndrome mortality is not accurate, however, a review with 58 cases found a mortality rate of 34% and no association between the deaths and the response to the treatment6. Additionally, the literature shows only one case of spontaneous improvement16.
In the past, a very low response to immunotherapy was reported in the first cases. However, new studies have been published describing a good response to immunotherapy- which can be explained by the inclusion of patients with non-classical phenotypes. Also, a higher response rate in patients with cognitive impairment and non-classical phenotypes happened in patients without HLA-DRB*1001 and cerebral spinal fluid inflammation. A systematic review showed that combination therapy can be more effective than monotherapy. Monotherapy response to immunotherapy was described in 7/22 patients (31,8%) while with combination therapy there was a response in 14/21 (66,6%)16.
The possible benefits of an immediate immunotherapy are an early antibody production interruption - resulting in a delay or stopping of the pathomechanism-, and a IgLON5 antibodies reduction8. Although, the reason why some patients are more responsive to immunotherapy is not clear and possibly had relations with the different phenotypes17.
Conclusions
Therefore, the Anti-IgLON5 syndrome is a disease with high mortality and several clinical manifestations. The pathophysiology of this condition is still unclear, but it is known there is an IgLON5 antibody involvement an important biomarker of the disease, and there is hyperphosphorylated Tau protein accumulation mainly in the hypothalamus and brain stem tegmentum. The analysis of symptoms, serum and CSF samples, alleles (HLADRB1*1001 and HLA-DQB1*0501), and the p-Tau protein presence allows the patient to be categorized as "definitive", "probable" and "possible" diagnosis. Besides, patients with Anti-IgLON5 disease may respond to immunotherapy, however, it depends on some clinical and analytical characteristics. Finally, future studies are needed to thoroughly study each particularity of this important pathological syndrome.
Summary
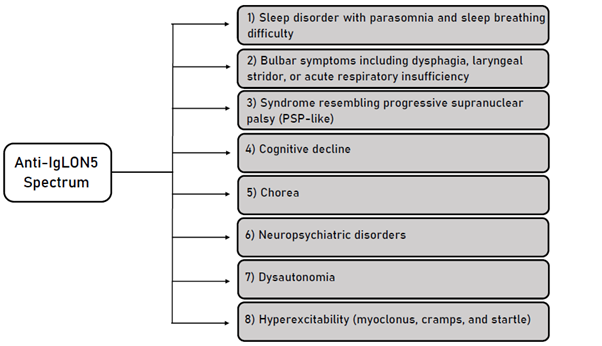
Figure 1: Summary of Anti- IgLON5 Spectrum of clinical manifestations.
Disclosure
The authors report no disclosures. All authors and contributors agree to the conditions outlined in the Authorship and Contributorship section of the Information for Author. The authors have read the Journal’s position on issues involved in ethical publication. There was no sponsorship for the scientific article and there was no conflict of interest with all the authors.
References
- Wenninger S. Expanding the Clinical Spectrum of IgLON5-Syndrome. Journal of Neuromuscular Diseases. 2017; 4(4): 337-339.
- Montagna M, Amir R, De Volder I, et al. IgLOn5- Associated Encephalitis with atypical brain magnetic resonance imaging and cerebrospinal fluid changes. Frontiers in Neurology. 2018; 9: 329.
- Karagogeos D. Neural GPI-anchored cell adhesionmolecules. Front Biosci. 2003; 8: s1304–20.
- Heidbreder P. Anti- Ig LON5 Disease. Curr Treat Option Neurol. 2018; 20(8): 29.
- Kubick N, Brosamle D, Mickael ME. Molecular evolution and functional divergence of the IgLON family. Evol Bioinformatics Online. 2018; 21(14):1176934318775081.
- Nissen MS, Blaabjerg M. Anti-IgLON5 Disease: A Case With 11-Year Clinical Course and Review of the Literature. 2019; 10: 1–7.
- Gaig C, Graus F, Compta Y, et al. Clinical manifestations of the anti-IgLON5 disease. Neurology. 2017; 88(18): 1736-1743.
- Sabater L, Planaguma, J, Dalmau J, et al. Cellular investigations with human antibodies associated with the anti-IgLON5 syndrome. Journal of Neuroinflammation. 2016; 13(1). doi:10.1186/s12974-016-0689-1.
- Gelpi E, Hoftberger R, Graus F, et al. Neuropathological criteria of anti-IgLON5-related tauopathy. Acta Neuropathologica. 2016; 132(4): 531-543.
- Sabater L, Gaig C, Gelpi E, et al. A novel non-rapid-eye movement and rapid-eye-movement parasomnia with sleep breathing disorder associated with antibodies to IgLON5: a case series, characterisation of the antigen, and post-mortem study. Lancet Neurol. 2014; 13: 575–586.
- Iranzo A. Parasomnias and sleep-related movement disorders in older adults. Sleep Med Clin. 2018; 13(1): 51–61.
- Honorat J, Komorowski L, Josephs KA, et al. IgLON5 antibody- Neurological accompaniments and outcomes in 20 patients. Neurology - Neuroimmunology Neuroinflammation. 2017; 4(5): e385.
- Schroder J, Melzer N, Ruck T, et al. Isolated dysphagia as initial sign of anti-IgLON5 syndrome. Neurology - Neuroimmunology Neuroinflammation. 2016; 4(1): e302.
- Graus F, Santamaria J. Understanding anti-IgLON5 disease. Neurol Neuroimmunol Neuroinflamm. 2017; 4: e393.
- Morales-Briceno H, Cruse B, Fois AF, et al. IgLON5-mediated neurodegeneration is a differential diagnosis of CNS Whipple disease. Neurology. 2018; 90(24): 1113-1115.
- Cabezudo-Garcia P, Mena-Vazquez N, Estivill Torrus G, et al. Response to immunotherapy in anti-IgLON5 disease: a systematic review. Acta Neurol Scand. 2020; 1(4): 263-270.
- Logmin K, Moldovan AS, Elben S, et al. Intravenous immunoglobulins as first-line therapy for IgLON5 encephalopathy. 2019. https://doi.org/10.1007/s00415-019-09221-3