
Brilaroxazine (RP5063) Clinical Experience in Schizophrenia: "A New Option to Address Unmet Needs"
Laxminarayan Bhat*, Marc Cantillon, and Robert Ings
Reviva Pharmaceuticals, Inc., Sunnyvale, CA, USA
Abstract
Schizophrenia is a condition comprising of both treatment and comorbidity factors that both complicate its management and present multiple unmet needs. Brilaroxazine (RP5063), a dopamine (D)/serotonin (5-HT) modulator, possesses a broad in vitro pharmacology profile against D2/3/4 and 5-HT1A/2A/2B/6/7 receptors, nicotinic acetylcholine (α4β2) receptors, and the serotonin transporter. In Phase 1 and 2 clinical experience in healthy volunteers, patients with schizophrenia and schizoaffective disorder, brilaroxazine was well tolerated, with the repeated 100 mg oral dose as the maximum tolerated dose. Investigators observed no cardiometabolic, cardiovascular, prolactin, or neurologic complications. Adherence in Phase 2 was good with discontinuation rates generally less than placebo. In a Phase 2 evaluation of patients with acute exacerbations in schizophrenia and schizoaffective disorders, brilaroxazine met its primary endpoint of significance versus placebo for Total Positive and Negative Symptom Scale (PANSS) Score at Day 28 as compared to baseline. In Phase 1 multi-dose pharmacodynamic evaluation, brilaroxazine displayed clinical activity in patients with stable disease and a PANSS greater than 50, as early as Day 10 of treatment. Pharmacokinetic analyses demonstrated brilaroxazine to have a substantial and relatively rapid oral absorption, linear (dose proportional) increases in maximum concentration (Cmax) and area under the curve (AUC), lack of excessive accumulation, and a relatively long terminal half-life of about 60 hours. Early clinical findings with brilaroxazine in schizophrenia and schizoaffective disorders indicate that this compound appears to be effective, well tolerated, and possess a straightforward once-daily dosing pharmacokinetic profile.
Introduction
Schizophrenia is a chronic and debilitating psychiatric syndrome affecting 1% of the world’s population1. It is a complex disease comprised of a mix of positive and negative symptoms, along with mood disorder and cognitive impairment2-4. Interestingly, psychosis is also linked to several neurodegenerative disorders (NDDs), including Alzheimer disease, spectrum of Parkinson's disease, and Huntington’s disease5-8.
Current pharmacologic treatment includes typical and atypical agents9. Tolerability issues (e.g., neuroleptic side effects with typical antipsychotics; metabolic and cardiovascular problems with atypical medications) limit compliance and effectiveness of both classes10-13. Hence, compliance is poor, with discontinuation rates of 30-50% in the short-term management of acute patients and 42-74% in the long-term treatment9,14-22. Also, both classes of antipsychotics fail to provide a broad spectrum of efficacy across the various symptom classes23-24. It is not surprising, therefore, that 30% of patients remain refractory to current treatments and critical needs remain to be met in the management of schizophrenia25.
Brilaroxazine (RP5063), a promising therapeutic presently under clinical development, possesses a broad in vitro pharmacology profile against dopamine (D) and serotonin (5-HT) receptors that underlie the pathogenesis of schizophrenia and related neuropsychiatric disorders. This compound possesses a high binding affinity for the D2/3/4 and 5-HT1A/2A/2B/7 receptors and moderate binding affinity for the 5-HT6 and nicotinic acetylcholine (α4β2) receptors, along with the serotonin transporter26,27. It displays partial agonist activity at the D2/3/4 and 5-HT1A/2A receptors, and antagonist activity at the 5-HT2B/6/7 receptors26.
Preclinical studies have established the activity, pharmacokinetics, and safety profile of brilaroxazine in animals (unpublished). Rodent models of pharmacologic-induced behaviors associated with schizophrenia have demonstrated that brilaroxazine limits both psychosis and cognitive symptoms28,29.
Considering the preclinical profile and unmet needs with present treatment options, Reviva Pharmaceuticals, Inc., proceeded to develop brilaroxazine for schizophrenia and schizoaffective disorders. Over this past year, investigators published four clinical research papers that describe the Phase 1 and 2 experience with this compound. The following discussion examines the safety, efficacy, pharmacokinetic, and pharmacodynamic results seen in Phase 1 studies (single-dose and multiple doses) and the Phase 2 trial, along with their implications.
Phase 1 Studies Provide a Promising Safety, Pharmacokinetic, and Pharmacodynamic Profile
Two Phase 1 studies were undertaken to define the initial clinical experience30,31. The first-in-human study involved an ascending single-dose study, and the first-in-patient study involved a multiple-dose ascending study over 10 days. Both studies characterized the initial safety and pharmacokinetic profiles in normal healthy volunteers (Caucasian or Japanese men, 20–45 years, 18.5–30.0 kg/m2) and in stable patients with schizophrenia (18–65 years, 19.5–38.0 kg/m2, chronic, all types with Total Positive and Negative Syndrome Scale [PANSS] score < 90 points). Also, the multiple-dose study provided initial insight into the pharmacodynamic effects in stable patients.
The single-dose was a dose-escalation study recruiting 24 individuals, with 23 completing the full evaluation. Initially, it examined patient cohorts receiving individual doses of 10 and 15 mg fasting, followed by a food-effect group (food versus fasting, crossover) with 15 mg dose. The multiple-dose study examined multiple doses of 10, 20, 50, and 100 mg given with food over ten days in 32 randomized patients.
Safety
In each study, brilaroxazine displayed an encouraging safety profile (Tables 1)30,31. In the single-dose study, 32 treatment-emergent adverse events (TEAEs) were observed, with orthostatic hypotension (n = 6), nausea (n = 4), and dizziness (n = 4) being the most common. Neither food nor race influenced the safety results. In the multiple-dose study, brilaroxazine was well tolerated at doses from 10 to 100 mg. Seventy-five TEAEs were reported, with somnolence (n = 16) and akathisia (n =12) the most frequent. Two serious adverse events (akathisia) were reported only at the 100-mg dose. These studies did not see any clinically significant changes in glucose or prolactin levels, lipid profiles, weight, or electrocardiographic recordings. In both studies, all TEAEs resolved, and none led to withdrawal or death.
Table 1: TEAEs by Preferred Term in Phase 1 and 2 Studies of Brilaroxazine26,30,31.
| Phase 1 Single-Dose Ascending Study (> 10% of Patients for Total) | ||||||
| TEAE by Preferred Term | Placebo (N = 4) |
10 mg (N = 6) |
15 mg (N = 6) |
Fed 15 mg (N = 7) |
Fast 15 mg (N = 8) |
Total brilaroxazine |
| Orthostatic | 0 (0.00) | 0 (0.00) | 1 (16.67) | 1 (14.29) | 4 (50.00) | 6 (22.22) |
| Nausea | 0 (0.00) | 1 (16.67) | 2 (33.33) | 0 (0.00) | 1 (12.50) | 4 (14.81) |
| Dizziness | 0 (0.00) | 0 (0.00) | 1 (16.67) | 1 (14.29) | 2 (25.00) | 4 (14.81) |
| Tachycardia | 0 (0.00) | 0 (0.00) | 1 (16.67) | 0 (0.00) | 1 (12.50) | 2 (7.40) |
| Fatigue | 0 (0.00) | 2 (33.33) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 2 (7.40) |
| Hyperhidrosis | 0 (0.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 2 (25.00) | 2 (7.40) |
| Phase 1 Multi-Dose Ascending Study (10 Days, Fed) (> 5% of Patients for Total) | ||||||
| TEAE by Preferred Term | Placebo (N = 8) |
10 mg (N = 6) |
20 mg (N = 6) |
50 mg (N = 6) |
100 mg (N = 6) |
Total brilaroxazine |
| Somnolence | 3 (37.50) | 2 (33.33) | 4 (66.67) | 5 (83.33) | 5 (83.33) | 16 (66.67) |
| Akathisia | 0 (0.00) | 0 (0.00) | 3 (50.00) | 3 (50.00) | 6 (100.00) | 12 (50.00) |
| Gastrointestinal | 0 (0.00) | 0 (0.00) | 1 (16.67) | 4(66.66) | 2 (33.33) | 7 (29.16) |
| Nausea | 0 (0.00) | 0 (0.00) | 1 (16.67) | 1 (16.67) | 1 (16.67) | 3 (12.50) |
| Extrapyramidal | 0 (0.00) | 1 (16.67) | 0 (0.00) | 1 (16.67) | 1 (16.67) | 3 (12.50) |
| Psychotic Disorder | 0 (0.00) | 1 (16.67) | 1 (16.67) | 1 (16.67) | 0 (0.00) | 3 (12.50) |
| Insomnia | 1 (12.50) | 1 (16.67) | 1 (16.67) | 1 (16.67) | 0 (0.00) | 3 (12.50) |
| Dystonia | 0 (0.00) | 0 (0.00) | 0 (0.00) | 2 (33.33) | 0 (0.00) | 3 (12.50) |
| Constipation | 0 (0.00) | 0 (0.00) | 0 (0.00) | 2 (33.33 | 0 (0.00) | 3 (12.50) |
| Vomiting | 0 (0.00) | 0 (0.00) | 0 (0.00) | 1 (16.67) | 1 (16.67) | 2 (8.33) |
| Phase 2 Study (28 Days, Fed) (> 5% of Patients for Total) | ||||||
| TEAE by Preferred Term | Placebo (N =38) |
15 mg (N = 58) |
30 mg (N = 59) |
50 mg (N = 58) |
Aripiprazole (N = 20) |
Total brilaroxazine |
| Insomnia | 8 (21.00) | 16 (28.00) | 15 (25.00) | 10 (17.00) | 41 (23.00) | 41(23.42%) |
| Agitation | 6 (16.00) | 6 (10.00) | 6 (10.00) | 4 (7.00) | 16 (9.00) | 22 (12.57%) |
| Extrapyramidal Disorder | 0 (0.00) | 2 (3.00) | 3 (5.00) | 5 (9.00) | 1 (5.00) | 10 (5.71) |
| Akathisia | 0 (0.00) | 1 (2.00) | 3 (5.00) | 6 (10.00) | 1 (5.00) | 10 (5.71) |
N = number of volunteers receiving at least 1 dose; n = number of volunteers with AEs. TEAE= treatment-emergent adverse events. Percentages were calculated using the number of volunteers receiving at least 1 dose (N) in each cohort as the denominator.
Adapted with permission from [Elsevier]: [Elsevier] [Schizophrenia Research] [REFERENCE 26 (Dopamine serotonin stabilizer RP5063: A randomized, double-blind, placebo-controlled multicenter trial of safety and efficacy in exacerbation of schizophrenia or schizoaffective disorder; Cantillon M, Ings R, Prakash A, and Bhat L), [COPYRIGHT] (2017) Adapted with permission from [John Wiley & Sons]: [John Wiley & Sons] [Clinical Translational Science] [REFERENCE 30 (Initial Clinical Experience of RP5063 Following Single Doses in Normal Healthy Volunteers and Multiple Doses in Patients with Stable Schizophrenia; Cantillon M, Ings R, Prakash A, and Bhat L), [COPYRIGHT] (2018).
Pharmacokinetics
These studies provided for an initial assessment of the pharmacokinetic profile (Table 2, Figures 1 and 2)30,31. In the single-dose study, plasma concentrations of brilaroxazine peaked at about 5 hours after dosing and then declined in a biphasic manner with a terminal half-life of approximately 40 to 50 hours. Mean Cmax and area under the curve (AUC)∞ increased in a linear, dose-proportional fashion over the dosing range. Food did provide a slight increase in drug absorption as seen in Cmax (reaching a median Tmax of 6 hours post-dose), and AUC∞, representative of drug exposure. Neither race (Japanese versus Caucasian) nor CYP2D6 genotype appear to influence the pharmacokinetic profile.
Table 2: Pharmacokinetic Parameters from Phase 1 Single- and Multiple-dose Ascending Studies of Brilaroxazine26,30,31.
| Single-Dose Ascending Study | |||||
| Pharmacokinetic Parameter | Unit Statistic | Cohort 1: 10 mg (n = 6) |
Cohort 2: 15 mg (n = 6) |
Cohort 3: (Food Effect) 15 mg Fed (n = 7) Fast (n = 8) |
|
| Cmax (ng/mL) | Mean (CV%) | 27.9 (34.0) | 36.6 (25.2) | 37.0 (33.0) | 34.0 (29.1) |
| Tmax (hr) | Median (min-max) | 5.0 (2.0-6.0) | 5.0 (2.0-6.0) | 6.0 (4.0-30.0) | 6.0 (1.5-8.0) |
| AUClast | Mean (CV%) | 565.4 (27.0) | 1170.1 (32.7) | 1848.2 (51.3) | 1544.1 (55.7) |
| AUCinf (hr*ng/mL) | Mean (CV%) | 1040.1 (39.1) | 1447.8 (35.2) | 2441.1 (71.2) | 1897.8 (67.7) |
| Cl/F (mL/hr) | Mean (CV%) | 10713.1 | 11496.2 (34.2) | 8886.2 (60.9) | 11118.7 (58.0) |
| T½ (hr) | Mean (CV%) | 44.8 (28.8) | 41.9 (24.4) | 56.4 (52.6) | 52.9 (36.1) |
| Multi-Dose Ascending Study | |||||
| Pharmacokinetic Parameter | Unit Statistic | Cohort 1: 10 mg (n = 6) |
Cohort 2: 20 mg (n = 6) |
Cohort 3: 50 mg (n = 6) |
Cohort 4: 100 mg (n = 6) |
| Cmax (ng/mL) 1 min | Mean (CV%) | 20.2 (18.87) | 43.0 (31.2) | 106.5 (20.7) | 195.3 (26.7) |
| Cmax (ng/mL) 10 min | Mean (CV%) | 70.1 (25.41) | 140.7 (27.5) | 292.2 (41.4) | 696.4 (43.3) |
| Tmax (hr) 1 min | Median (min-max) | 5.0 (2.0-6.0) | 5.0 (2.0-6.0) | 6.0 (4.0-30.0) | 6.0 (1.5-8.0) |
| Tmax (hr) 10 min | Median (min-max) | 6.0 (4.0-12.0) | 6.0 (1.5-8.0) | 6.0 (4.0-16.0) | 6.0 (4.0-12.0) |
| AUClast (hr*ng/mL) | Mean (CV%) | 565.4 (27.0) | 1170.1 (32.7) | 1848.2 (51.3) | 1544.1 (55.7) |
| AUClast (hr*ng/mL) | Mean (CV%) | 4576.2 (50.9) | 7991.5 (43.5) | 21337.7 | 48805.6 (58.1) |
| T½ (hr) | Mean (CV%) | 68.1 (47.0) | 55.1 (22.4) | 70.9 (23.9) | 58.8 (35.2) |
AUC: Area under the curve; Cmax: Maximum concentration; CV: Coefficient of variation as a percentage. AUC: Area under the curve; CL/F: Clearance; Cmax: Maximum concentration; hr: Hours; Tmax: Time of maximum concentration; T½ = half-life. * Tmax is described by median and range.
Adapted with permission from [John Wiley & Sons]: [John Wiley & Sons] [Clinical Translational Science] [REFERENCE 31 (Pharmacokinetics of RP5063 Following Single Doses to Normal Healthy Volunteers and Multiple Doses Over 10 Days to Stable Schizophrenic Patients; Cantillon M, Ings R, Prakash A, and Bhat L), [COPYRIGHT] (2017).
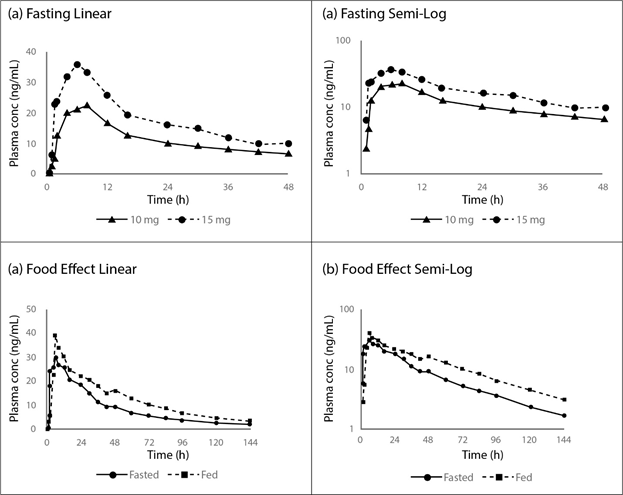
Figure 1: Concentration-Time Profiles for Fasting (a) Linear and (b) Semi-Log, and Food Effect (c) Linear and (d) Semi-Log from the Phase 1 Single-dose Study of Brilaroxazine in Normal Healthy Volunteers30.
Data reflect rapid and dose-dependent absorption, along with a slight effect of food, with single doses. Reprinted with permission from [John Wiley & Sons]: [John Wiley & Sons] [Clinical Translational Science] [REFERENCE 31 (Pharmacokinetics of RP5063 Following Single Doses to Normal Healthy Volunteers and Multiple Doses Over 10 Days to Stable Schizophrenic Patients; Cantillon M, Ings R, and Bhat L), [COPYRIGHT] (2017).
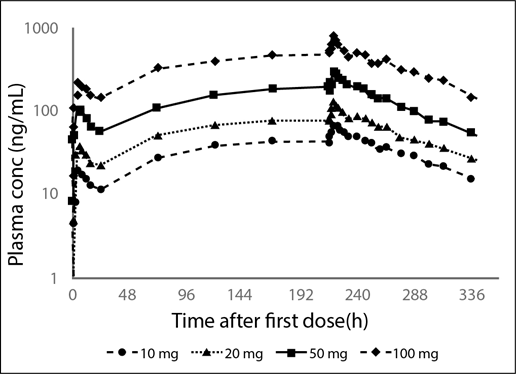
Figure 2: Mean Plasma Profiles from the Phase 1 Multiple Dose Ascending Data of Brilaroxazine when Administered Daily at Increasing Oral Doses for 10 Days to Stable Patients with Schizophrenia30.
Data reflect the dose-dependent plasma concentrations with ascending multiple doses, reaching of steady state at 120 hours, and limited accumulation
Reprinted with permission from [John Wiley & Sons]: [John Wiley & Sons] [Clinical Translational Science] [REFERENCE 31 (Pharmacokinetics of RP5063 Following Single Doses to Normal Healthy Volunteers and Multiple Doses Over 10 Days to Stable Schizophrenic Patients; Cantillon M, Ings R, and Bhat L), [COPYRIGHT] (2017).
In the multiple-dose study, Cmax and AUCss also increased in a linear, dose-proportional manner from 10 to 100 mg, (20.2 ng/mL to 195.0 ng/mL, respectively for Cmax and from 314.0 hour*ng/mL to 3538.0 hour*ng/mL, respectively for AUCss). Mean Cmax was reached at a median time of 4 to 5 hours post-dose across all dose levels. Mean trough concentrations indicate that by 120 hours of daily dosing steady-state was approached. The half-life (ranging from 55 to 71 hours) was similar across all the dose levels.
Pharmacodynamic Assessment
Since the multiple-dose study was conducted in stable patients with schizophrenia, an initial pharmacodynamic evaluation was undertaken to provide insight into the clinical activity of brilaroxazine31. While brilaroxazine (pooled doses) did not show significance versus placebo in PANSS Total Score or individual subscales specific to change from baseline for each visit, it did show promising signals of activity in several secondary analyses. These included significant improvements (P<0.05) versus placebo in (1) PANSS Marder-derived Depression/Anxiety and Impulsivity/Hostility Factors and (2) PANSS secondary analysis for Positive Subscale Scores of patients with a baseline PANSS score of 50 or higher (Figure 3)31-33. Furthermore, improvements in Trails A and Trails B Test results were observed for patients treated in the 50 mg dose group (Days 5, 10, and 16).
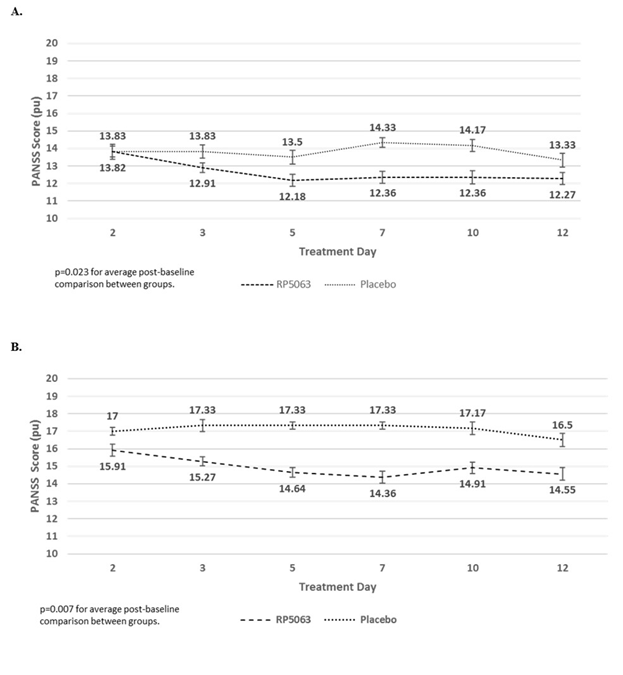
Figure 3: PANSS Repeated Measures Analysis of Patients with PANSS >50 from the Phase 1 Multiple-dose Ascending Study for (A) Positive Subscale Scores and (B) Positive Subscale Scores with Marder Factors for Brilaroxazine (Pooled Data, All Doses), as Compared with Placebo31.
Data reflect an improvement in effect on reducing PANSS positive subscale scores (A) and PANSS positive subscale scores with Marder factors (B) in stable patients with schizophrenia who present with a baseline PANSS >50 treated with brilaroxazine in the Phase 1 multidose ascending study. The average post-baseline effect, as compared with placebo, was significant with brilaroxazine in both assessments (P=0.023 for PANSS positive subscale scores, P=0.007 for PANSS positive subscale scores with Marder factors).
Reprinted with permission from [John Wiley & Sons]: [John Wiley & Sons] [Clinical Translational Science] [REFERENCE 30 (Initial Clinical Experience of RP5063 Following Single Doses in Normal Healthy Volunteers and Multiple Doses in Patients with Stable Schizophrenia; Cantillon M, Ings R, Prakash A, and Bhat L), [COPYRIGHT] (2018).
However, since treatment was for 10-days and in stable patients, it was not surprising that inconsistencies in its effect across doses for Clinical Global Improvement (CGI) and Bond and Lader visual analog scales precluded any meaningful conclusions from these determinations.
Phase 2 Trial Adds to the Safety, Efficacy and Pharmacokinetics/Pharmacodynamic Picture
The Phase 2 (REFRESH; NCT01490086) evaluation was conducted in patients with acute exacerbations of schizophrenia or schizoaffective disorder to evaluate the efficacy, safety, tolerability, and pharmacokinetics of brilaroxazine versus placebo26. This 28-day, multicenter, placebo-controlled, double-blind study randomized 234 subjects to brilaroxazine (15, 30, or 50 mg); aripiprazole (15 mg); or placebo (ratio, 3:3:3:1:2) once daily. The primary endpoint was changed from baseline to Day 28/End-of-Treatment of PANSS total score, a standardized, validated drug sensitive instrument that is commonly used in registration trials to assess positive, negative, and cognitive symptoms in patients with schizophrenia3. For this analysis, the total PANSS measurements for those patients dosed with brilaroxazine were used as a response factor. Study participants were dosed daily following an overnight fast before breakfast for 28 days and followed up 1 week after the last dose. Patients were required to remain in the testing facility for the study duration to ensure accurate dosing and sample time collection. Safety analysis involved the collection and analysis of adverse events, laboratory, and discontinuation rates. Plasma samples were obtained from patients treated with brilaroxazine.
Clinical Efficacy
The primary analysis of PANSS Total (Figure 4) showed improvement with brilaroxazine in the 15, 30, and 50 mg arms with a mean (SE) of −20.23 (2.65) −15.42 (2.04), and −19.21 (2.39), respectively. The difference between treatment and placebo reached statistical significance for the 15 mg (P = 0.021) and 50 mg (P= 0.016) arms. CGI-Severity improvement of ≥2 points by Day 28 was observed at twice the frequency of placebo (P<0.05 for 15- and 50-mg doses versus placebo). Numerical improvements for cognition were seen in the Digital Symbol Substitution Test, and Trails A and Trails B scores.
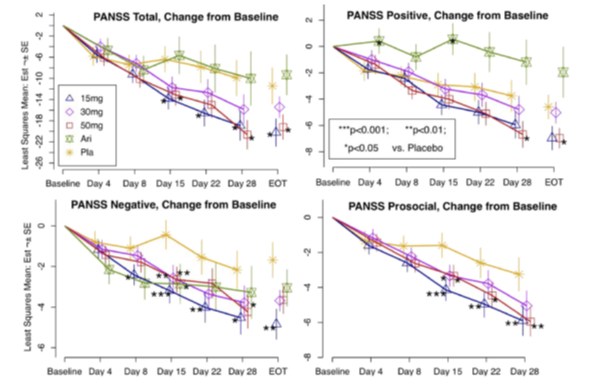
Figure 4: Change from Baseline PANSS Total, Positive, Negative and Social Subscales Observed in the Phase 2 Study of Brilaroxazine in Patients with Acute Schizophrenia and Schizoaffective Disorder26.
Day 4 to Day 28 are LSMs with standard errors (SEs) and P-values from the mixed-effects model repeated measures (MMRM) analysis, including treatment, baseline, and region as fixed effects, and site, subject and subject time trend as random effects. End-of-Treatment (EOT) estimates are the Day 28/EOT values with standard errors and P-values from the Mann-Whitney U test for differences between treatment groups.
Reprinted by permission from [Elsevier]: [Elsevier] [Schizophrenia Research] [REFERENCE 26 (Dopamine serotonin stabilizer RP5063: A randomized, double-blind, placebo-controlled multicenter trial of safety and efficacy in exacerbation of schizophrenia or schizoaffective disorder; Cantillon M, Ings R, Prakash A, and Bhat L), [COPYRIGHT] (2017).
Safety
The most common TEAEs for brilaroxazine were insomnia (23.42%) and agitation (12.57%). Body weight, electrocardiogram, or incidence of orthostatic hypotension did not display any significant changes. Interestingly, blood glucose, lipid profiles, and prolactin levels all decreased rather than increased, unlike that seen with other atypical psychotropics. Discontinuation for any reason for brilaroxazine (14%, 25%, 12%) was much lower than for placebo (26%) and aripiprazole (35%). There were no side effects related dropouts in brilaroxazine dose groups 15 mg and 30 mg. The highest dose (50mg) of brilaroxazine showed only 2% side effect related dropouts, whereas aripiprazole 15 mg showed 10%.
Pharmacokinetic and Pharmacodynamic Analyses and Models
Analysis of Phase 2 trial data involving patients with schizophrenia and schizoaffective disorder was undertaken to define: (1) the pharmacokinetics of brilaroxazine; and (2) the pharmacokinetic/ pharmacodynamic relationships of brilaroxazine utilizing the improvement in the primary endpoint (Total PANSS)34. This analysis was performed to complement the pharmacokinetic profile and to provide useful data to guide dose planning in patients with acute exacerbations of schizophrenia or schizoaffective disorder for the Phase 3 trial.
During the Phase 2 trial, five blood samples (10 mL) were collected from each patient pre-dose at baseline and at four other time intervals, using sampling blocks of time with a predesignated number of patients per block, so samples were obtained encompassing the whole dosing period up to 220 hours after the last dose26. Additional data collected, included covariates (body weight, body mass index, age, sex, smoking, concomitant drug use, race/ethnicity, geographic area of the clinical site, and creatinine clearance [Cockcroft Gault] as a surrogate for glomerular filtration rate. The population pharmacokinetic analysis involved the development of 1- and 2-compartment models and the evaluation of the impact of co-variates utilizing bootstrap techniques. The population pharmacodynamic analysis included the development of an Emax model for total PANSS from the Phase 2 trial as the response factor against cumulative AUC34.
The pharmacokinetic analysis identified a 1-compartmental model incorporating the covariate of body mass index as the optimum construct for this sparse data set (Table 3). The fixed-effect parameters were, oral clearance (Cl/F), 5.11 ± 0.11 L/h and volume of distribution (V/F), 328.00 ± 31.40 L; absorption constant (ka) 0.41 ± 0.17 h-1; and lag time (t-lag) of 0.41 ± 0.02. A statistical evaluation showed that the brilaroxazine pharmacokinetic profile was linear. The calculated half-life was 44.5 hours like that seen in the earlier studies. Thus, the parameters predicted from this analysis were consistent with those obtained previously in normal healthy volunteers and patients with stable schizophrenia using a traditional pharmacokinetic approach (Cantillon et al., 2017[a]).
Table 3: Final Population Pharmacokinetic Model Parameter Estimates Derived from the Stepwise Covariate Analysis of Phase 2 Study Data of Brilaroxazine in Patients with Schizophrenia or Schizoaffective Disorder34.
| Parameter | Estimate | Standard Error (SE) | Bootstrap | Bootstrap SE |
|---|---|---|---|---|
| Cl (L/h) | 5.11 | 0.11 | 5.11 | 0.17 |
| Cl (L/h) | 328.00 | 31.4 | 329 | 2.05 |
| ka (h-1) | 0.42 | 0.17 | 0.45 | 0.12 |
| t-lag (h) | 0.41 | 0.02 | 0.47 | 0.11 |
| γ γ | 0.90 | 0.36 | 0.84 | 0.34 |
| var(η1) | 0.16 | 0.10 | 0.16 | 0.02 |
| var(η2) | 0.28 | 0.16 | 0.256 | 0.07 |
| Cov (η1, η2) | 0.04 | 0.15 | 0.05 | 0.03 |
| var (η3) | 2.09 | 6.63 | 2.36 | 0.66 |
| σ12 | 0.07 | 0.02 | 0.07 | 0.001 |
Cl = Clearance; Cov=covariance; γ= Parameter that quantifies the extent of the relationship among covariates; ka= Absorption rate constant; η1= Random effects and are multivariate normal with mean zero and variance-covariance matrix Ω; σ12 = Variance characterizing the intrasubject variability; t-lag=lag time; Var= Variance; V = Volume of distribution
Reprinted by permission from [Springer]: [Springer] [European Journal of Drug Metabolism and Pharmacokinetics] [REFERENCE 34 (A Population Pharmacokinetic and Pharmacodynamic Analysis of RP5063 Phase 2 Study Data in Patients with Schizophrenia or Schizoaffective Disorder; Cantillon M, Ings R, Prakash A, and Bhat L), [COPYRIGHT] (2018).
An Emax model (Table 4) for total PANSS as the response factor against cumulative AUC was identified as the best construct that demonstrated high predictability and low variability when correlated with actual observations. This model provided fixed effect estimates for E0 of 87.3±0.71 PANSS Units (pu), Emax-31.60 ±4.05 (pu) and AUC50 89.60±30.10 (µg*h/mL). The predicted PANSS improvement indicated an adequate clinical dose range of between 5 and 30 mg (Figure 5).
Table 4: Final Emax Model Parameter Estimates Incorporating Baseline and Site as Identified from the Population Covariate Analysis of Data from the Phase 2 Study of Brilaroxazine in Patients with Acute Schizophrenia or Schizoaffective Disorder34.
| Parameter | Estimate | Standard Error (SE) |
|---|---|---|
| E0 (pu) | 87.3 | 0.711 |
| Emax (pu) | -31.6 | 4.05 |
| AUC50 (μg*h/mL) | 89.6 | 30.1 |
| γ | -1.93 | 0.535 |
| var (η1) | 164 | 632 |
| var (η2) | 464 | 308 |
| var (η3) | 0.476 | .878 |
| σ2 | 47.1 | 6.04 |
AUC50 = Area under the curve at 50% of maximal effect; Emax= The maximal effect at high drug concentrations when all the receptors are occupied by the drug; E0 = Baseline effect; γ = Parameter that quantifies the extent of the relationship. In this case, it relates to the influence of Geographic Area 5; ηj = = Random effects and are multivariate normal with mean zero and variance-covariance matrix Ω; pu= PANSS units; σ2 = Variance characterizing the intra-subject variability.
Reprinted by permission from [Springer]: [Springer] [European Journal of Drug Metabolism and Pharmacokinetics] [REFERENCE 34 (A Population Pharmacokinetic and Pharmacodynamic Analysis of RP5063 Phase 2 Study Data in Patients with Schizophrenia or Schizoaffective Disorder; Cantillon M, Ings R, Prakash A, and Bhat L), [COPYRIGHT] (2018).
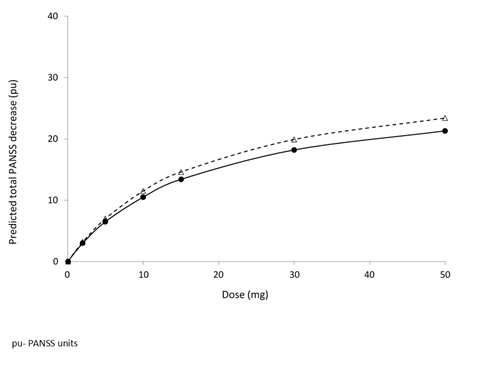
Figure 5: Relationship of Brilaroxazine as Measured by Total PANSS Based on Phase 2 Data from Patients with Schizophrenia and Schizoaffective Disorders34.
Solid line: base model; Dashed line: model incorporating the geographic site effect on Emax.
Data reflect dose response (PANSS) relationship to 30 mg, with subsequent diminishing response relationship at doses between 30 and 50 mg.
Reprinted by permission from [Springer]: [Springer] [European Journal of Drug Metabolism and Pharmacokinetics] [REFERENCE 34 (A Population Pharmacokinetic and Pharmacodynamic Analysis of RP5063 Phase 2 Study Data in Patients with Schizophrenia or Schizoaffective Disorder; Cantillon M, Ings R, Prakash A, and Bhat L), [COPYRIGHT] (2018).
Safety Profile Begins to Fulfill Unmet Needs for Schizophrenia
The initial Phase 1 and 2 studies with brilaroxazine reflect an encouraging safety profile26,30,31. It is essential to put this safety profile into perspective relative to the side-effect profiles of other available treatments, all of which can further complicate health status. When reviewing the data, several themes emerge relative to safety and tolerability.
Theme 1: Brilaroxazine Appeared to Be Well Tolerated
The overall incidence rates of TEAEs with brilaroxazine were low and more prevalent with higher doses (50 and 100 mg). These studies did not find any of these TEAEs were associated with any severe morbidity, mortality, or study withdrawals. Patient compliance was very favorable with discontinuation in the Phase 2 trial (12-25%), either lower or similar to placebo (26%) and lower than a positive control, aripiprazole (35%)26.
Theme 2: Issues Exist with Current Treatments that Warrant Newer Alternatives
When considering the two classes of current treatment, typical agents block dopamine receptors, mainly D2, leading to extrapyramidal symptoms and hyperprolactinemia11,13. Treatment with these antipsychotics can lead to a decrease in or loss of sexual interest directly through D2 receptor antagonism and indirectly through hyperprolactinemia10. These side effects can severely limit compliance, which has been reported as low as 40% to 50%11, 35,36. These agents can also contribute to unintentional weight gain37-39.
Atypical agents, on the other hand, exert antagonistic activity on 5-HT, specifically 5-HT2A, along with D2. These agents are also limited by their side effects13,40. Furthermore, atypical agents have been linked with weight gain and type 2 diabetes mellitus, leading to a US Food and Drug Administration warning in the package inserts of agents in this class31-43. Dibenzodiazepine-derived atypical agents (e.g., clozapine, olanzapine, and quetiapine) have also produced elevated serum triglyceride levels, with clozapine and olanzapine showing increased cholesterol levels after 8 weeks of treatment44,45. Direct cardiovascular effects have been noted including an increased QTc (ziprasidone), myocarditis (clozapine), and worsening hypertension control (aripiprazole)46-50. Finally, increased prolactin levels have been reported with amisulpride and risperidone, and retrograde ejaculation and priapism have been seen with clozapine, risperidone, and olanzapine1-13.
With brilaroxazine, metabolic parameters (e.g., blood glucose, lipid profiles, and prolactin) did not change significantly in these studies. There were no reports of sexual dysfunction. No significant changes were seen in body weight, clinical laboratories, electrocardiogram, or incidence of orthostatic hypotension, as compared with placebo, potentially providing a considerable advantage over currently existing medications.
Theme 3: Comorbidities Limit Clinician Chose and Define Further Unmet Needs
Furthermore, patients with schizophrenia experience multiple comorbidities at a higher rate than normal healthy individuals51. These factors include lifestyle (e.g., high smoking prevalence, high-fat diet), neglect of personal care, and barriers for treatment of ongoing conditions13. Between 40% and 60% of patients with schizophrenia are overweight, and their baseline cardiometabolic and cardiovascular risks are higher than those of the general population13,52. Psychosis is associated with reduced sexual interest, activity, and satisfaction53,54. Furthermore, direct cardiovascular effects present an even greater concern in older patients.
Additionally, between 30% and 50% of patients with NDDs (Alzheimer’s disease, Parkinson’s disease, Parkinson’s spectrum disorders, and Huntington’s disease) suffer from psychosis5-7,55. These neurological illnesses erode the patient’s cognition and motor skills, along with their very perception of reality, causing hallucinations and delusions, agitation and aggression, and other related phenomena56,57. The characteristics and severity of psychotic and behavioral symptoms in neurological diseases may differ from schizophrenia possibly due to more 5-HT dysfunction than dopamine55,58-61. Published experience involving NDDs with typical and atypical agents appear limited. Side effects from these treatments can potentially further complicate the neurologic condition, as well as contributing to poor compliance.
Hence, these underlying conditions can quickly limit clinician choice when considering the side-effect profiles of available medications and clearly define the need for newer alternatives62. The safety profile that displays no significant cardiovascular, cardiometabolic, laboratory, and neurologic effects may offer clinicians an alternative to fulfill present unmet needs related to patient safety and tolerability that limit persistency with therapy. This safety profile of brilaroxazine, along with its quick onset and efficacy profile, should enable the clinician to manage acute symptoms effectively without the need to switch to another drug for the long-term treatment of schizophrenia, where compliance can be poor.
Pharmacokinetic Profile Provides for a Straightforward Once-Daily Dosing Schedule
The parameters predicted from the population analysis were similar to those obtained from previous brilaroxazine Phase 1 studies in normal healthy volunteers and patients with stable schizophrenia using a traditional pharmacokinetic approach34. The results from these studies offer valuable approach into the direction of dosing regimen design in future stages of clinical investigation. When reviewing the data from these two papers, several key themes emerge from these studies regarding the pharmacokinetic profile of brilaroxazine.
Theme 1: The Pharmacokinetics of Brilaroxazine were Linear over the Complete Dose Range Anticipated for Clinical Use and in the Target Population.
Both the Phase 1 study data and Phase 2 analysis reflected linear, dose-proportional increases in exposure with dose and with no evidence of time dependency. Of significant note, the exposure of brilaroxazine as measured by Cmax and AUC increased in direct proportionality to drug dose irrespective of the population being used in the studies (healthy volunteers, stable schizophrenic patients or schizophrenic patients with acute exacerbations of schizophrenia or schizophrenic disorder).
Theme 2: The Lack of Accumulation Translates to No Need for Dose Titration.
In Phase 1 multi-dose study, drug levels approached steady-state after 120 h (5 days) of daily dosing, with doses between 10 and 100 mg with maximum steady-state concentrations of 70.1 and 696 ng/mL and AUCss of 1361 and 12526 ng*h/mL at the 10 and 100 mg dose, respectively. The lack of excessive accumulation translates to a potential clinical advantage of no need for titration of therapy, as may be the case with other atypical antipsychotics (e.g., aripiprazole)63.
Theme 3: The Long Half-life Translates to Once-Daily Dosing and Coverage of a Missed Dose
The half-life ranged from 40 h to 71.9 h from the three studies: (1) 40-50 h in single-dose; (2) 55-71.9 h in the multiple-dose; and (3) 44.5 h from the Phase 2 population modeling. Moreover, since brilaroxazine is a low clearance compound (Cl/F approx. 10% of hepatic blood flow), the similarity of the half-life between a single dose and after the 10th administration of a daily dosage regimen, together with AUCss predicted from single-dose data, would confirm the lack of time dependency. Nevertheless, the half-life of over 40 h translates easily to a once-daily dosing schedule, as well as being of clinical importance for the schizophrenic patient population, since medication adherence and missing doses of shorter half-life drugs can be a clinical issue leading to destabilization of clinical control14 and poor long-term functional outcomes in the treatment of schizophrenia. With brilaroxazine, if a patient misses a single dose or two, sufficient plasma concentrations remain for clinical control.
Theme 4: The Lack of Pharmacokinetic Differences Translates to Straightforward Dosing Across Populations
An examination of the pharmacokinetics of brilaroxazine from the studies revealed that the pharmacokinetics of brilaroxazine were independent of race, ethnicity, health status, and type of schizophrenia disorder treated. These observations mean that no dose adjustments are needed based on the patient population.
Conclusions
Both treatment and comorbidity factors complicate schizophrenia management. Safety, tolerability, breadth of symptom coverage, and simplicity of dosing limit current treatments. Furthermore, multiple comorbidities, including those with NDDs, make the clinician’s choice difficult. These all present numerous unmet medical needs.
The initial clinical experience of brilaroxazine in healthy volunteers and schizophrenia and schizoaffective disorder patients define this compound as a promising addition to the treatment armamentarium of this condition. Both healthy volunteers and patients tolerated brilaroxazine well in both Phase 1 and 2 studies. It did not produce any cardiometabolic, cardiovascular, prolactin, or neurologic side effects, which would complicate current treatments. Early activity was observed in results from the Phase 1 study after 10-days of dosing in stable patients and efficacy was seen in the findings from the Phase 2 trial, as brilaroxazine demonstrated significance versus placebo in Total PANSS Score at Day 28 as compared to baseline. The pharmacokinetics proved to be highly predictable and consistent between Phase 1 and 2 studies, participant type (healthy volunteer, patient), and racial characteristics (Black, Caucasian, Indian, and Japanese). Pharmacokinetic analyses showed substantive and relatively rapid oral absorption, linear, dose proportional increases in Cmax and AUC, lack of excessive accumulation, and a relatively long terminal half-life over 40 hours. These findings translate to a straightforward once-daily dosing regimen with no need for titration or adjustments based on racial characteristics. These characteristics set the stage for further evaluation in Phase 3, which is planned to start in soon.
Acknowledgments
Funding Support
These studies were supported by a grant to PAREXEL International and from Reviva Pharmaceuticals, Inc.
Phase 2 Pharmacokinetic/Pharmacodynamic Analysis was supported by a grant to Davide Verotta, Ph.D. at the School of Pharmacy, University of California, San Francisco, from Reviva Pharmaceuticals, Inc.
Editorial Support
Editorial support was provided by John M. York, PharmD, MBA. This assistance was funded by Reviva Pharmaceuticals, Inc.
Acknowledgement
Davide Verotta, Ph.D., of the School of Pharmacy, University of California, San Francisco, received grant funding to conduct the phase 2 population pharmacokinetic analysis and has nothing further to disclose.
Conflicts of Interest
Laxminarayan Bhat, Ph.D., and Marc Cantillon, MD, are employees of Reviva Pharmaceuticals, Inc. Robert Ings, Ph.D. is a consultant to Reviva Pharmaceuticals, Inc.
References
- van Os J, Kapur S. Schizophrenia. Lancet. 2009; 374(9690): 635–45.
- Lehman AF, Lieberman JA, Dixon LB, et al. American Psychiatric Association; Steering Committee on Practice Guidelines. Practice guideline for the treatment of patients with schizophrenia, Second edition. Am J Psychiatry. 2004; 161(2 Suppl): 1-56.
- Opler MGA, Yavorsky C, Daniel DG. Positive and Negative Syndrome Scale (PANSS) Training: Challenges, Solutions, and Future Directions. Innov Clin Neurosci. 2017; 14(11-12): 77-81.
- Stahl SM, Meghan M Grady. Stahl's Essential Psychopharmacology: The Prescriber's Guide. 4th ed. Cambridge, UK; New York: Cambridge University Press. 2011; 709.
- Kunik ME, Snow AL, Davila JA, et al. Consequences of Aggressive Behavior in Patients with Dementia. J Neuropsychiatry Clin Neurosci. 2010; 22(1): 40-47.
- Flynn FG, Cummings JL, Gornbein J. Delusions in dementia syndromes: investigation of behavioral and neuropsychological correlates. J Neuropsychiatry Clin Neurosci. 1991; 3(4): 364-370.
- Gilley DW, Wilson RS, Beckett LA, et al. Psychotic symptoms and physically aggressive behavior in Alzheimer’s disease. J Am Geriatr Soc. 1997; 45(9): 1074-1079.
- Ross CA, Margolis RL. Huntington’s disease. Clin Neurosci Res. 2001; 1(1): 142-152.
- Gobira PH, Ropke J, Aguiar DC, et al. Animal models for predicting the efficacy and side effects of antipsychotic drugs. Revista Brasileira de Psiquiatria. 2013; 35: S132–S139.
- Baldwin D, Mayers A. Sexual side-effects of antidepressant and antipsychotic drugs. Advances in Psychiatry Therapy. 2003; 9: 202-210.
- Hansen TE, Casey DE, Hoffman WF. Neuroleptic intolerance. Schizophr Bull. 1997; 23(4): 567-82.
- Meltzer HY, Risinger R, Nasrallah HA. A randomized, double-blind, placebo-controlled trial of aripiprazole lauroxil in acute exacerbation of schizophrenia. J Clin Psychiatry. 2015; 1085-90.
- Üçok A, Gaebel W. Side effects of atypical antipsychotics: a brief overview. World Psychiatry. 2008; 7: 58-62.
- Ascher-Svanum H, Faries DE, Zhu B, et al. Medication adherence and long-term functional outcomes in the treatment of schizophrenia in usual care. The Journal of Clinical Psychiatry. 2006; 67(3): 453-460.
- Cramer JA, Rosenheck R. Compliance with medication regimens for mental and physical disorders. Psychiatr Serv. 1998; 49(2): 196-201.
- Kahn RS, Fleischhacker WW, Boter H, et al. Effectiveness of antipsychotic drugs in first-episode schizophrenia and schizophreniform disorder: an open randomized clinical trial. Lancet. 2008; 371(9618): 1085-97.
- Kroken R.A, Kjelby E, Wentzel-Larsen T, et al. Time to discontinuation of antipsychotic drugs in a schizophrenia cohort: influence of current treatment strategies. Therapeutic Advances in Psychopharmacology. 2014; 4(6): 228–239.
- Komossa K, Rummel-Kluge C, Hunger H, et al. Risperidone versus other atypical antipsychotics for schizophrenia. Cochrane Database Syst Rev. 2010; (3): CD006654.
- Levine SZ, Goldberg Y, Samara M, et al. Joint modeling of dropout and outcome in three pivotal clinical trials of schizophrenia. Schizophr Res. 2015; 164(1-3): 122-6.
- Leucht S, Heres S, Hamann J, et al. Methodological issues in current antipsychotic drug trials. Schizophr Bull. 2008; 34(2): 275-85.
- Lieberman JA, Stroup TS, McEvoy JP, et al. Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) Investigators. N Engl J Med. 2005; 353(12): 1209-23.
- Potkin SG, Saha AR, Kujawa MJ, et al. Aripiprazole, an antipsychotic with a novel mechanism of action, and risperidone vs. placebo in patients with schizophrenia and schizoaffective disorder. Arch Gen Psychiatry. 2003; 60(7): 681-90.
- Rabinowitz J, Davidov O. A composite approach that includes dropout rates when analyzing efficacy data in clinical trials of antipsychotic medications. Schizophr Bull. 2008; 34(6): 1145-50.
- Kemmler G, Hummer M, Widschwendter C, et al. Dropout rates in placebo-controlled and active-control clinical trials of antipsychotic drugs: a meta-analysis. Arch Gen Psychiatry. 2005; 62(12): 1305-12.
- The American College of Neuropsychopharmacology (ACNP). Neuropsychopharmacology- 5th Generation of Progress (2010) Section 6, Schizophrenia. Available at: http://www.acnp.org/publications/neuro5thgeneration.aspx.
- Cantillon M, Prakash A, Alexander A, et al. Dopamine serotonin stabilizer RP5063: A randomized, double-blind, placebo-controlled multicenter trial of safety and efficacy in exacerbation of schizophrenia or schizoaffective disorder. Schizophr Res. 2017; 189: 126-133.
- Koester LS, Carbon M, Correll CU. Emerging drugs for schizophrenia: an update. Expert Opin. Emerg. Drugs. 2014; 19 (4): 511–531.
- Bhat L, Adiey K, Bhat S. RP5063, a novel, multimodal neuromodulator that stabilizes the dopamine-serotonin system and displays antipsychotic efficacy in rodent models of schizophrenia. Behavioural Pharmacology. 2018. (in review).
- Rajagopal L, Kwon S, Huang M, et al. RP5063, an atypical antipsychotic drug with a unique pharmacologic profile, improves declarative memory and psychosis in mouse models of schizophrenia. Behav Brain Res. 2017; 332: 180-199.
- Cantillon M, Ings R, Bhat L. Initial clinical experience of Rp5063 following single doses in normal healthy volunteers and multiple doses in patients with stable schizophrenia. Clin Transl Sci. 2018; 11(4): 387-396.
- Cantillon M, Ings R, Bhat L. Pharmacokinetics of RP5063 following single doses to normal healthy volunteers and multiple doses over 10 days to stable schizophrenic patients. Clin Transl Sci. 2017; 11(4): 378-386.
- Marder SR, Davis JM, Chouinard G. The effects of risperidone on the five dimensions of schizophrenia derived by factor analysis: combined results of the North American trials. J Clin Psychiatry. 1997; 58: 538–546.
- Marder SR, Essock SM, Miller AL, et al. The Mount Sinai conference on the pharmacotherapy of schizophrenia. Schizophr Bull. 2002; 28: 5-16.
- Cantillon M, Ings R, Prakash A, et al. A population pharmacokinetic and pharmacodynamic analysis of RP5063 phase 2 study data in patients with schizophrenia or schizoaffective disorder. Eur J Drug Metab Pharmacokinet. 2018: 43(5): 573-585.
- Birnbaum M, Sharif Z. Medication adherence in schizophrenia: patient perspectives and the clinical utility of paliperidone ER. Patient Prefer Adherence. 2008; 2: 233-40.
- Lacro JP, Dunn LB, Dolder CR, et al. Prevalence of and risk factors for medication nonadherence in patients with schizophrenia: a comprehensive review of recent literature. J Clin Psychiatry. 2002; 63(10): 892-909.
- Brown S. Excess mortality of schizophrenia. A meta-analysis Br J Psychiatry. 1997; 171: 502-508.
- Saddichha S, Manjunatha N, Ameen, et al. Effect of olanzapine, risperidone, and haloperidol treatment on weight and body mass index in first-episode schizophrenia patients in India: a randomized, double-blind, controlled, prospective study. J Clin Psychiatry. 2007; 68(11): 1793-8.
- Allison DB, Mentore JL, Heo M, et al. Antipsychotic-induced weight gain: a comprehensive research synthesis. Am J Psychiatry. 1999; 156: 1686-96.
- Panariello F, De Luca V, de Bartolomeis. New findings from animal model and pharmacogenomic studies. Schizophrenia Research and Treatment. 2011; 459284.
- Alvarez-Jimenez M, Gonzalez-Blanch C, et al. Attenuation of antipsychotic-induced weight gain with early behavioral intervention in drug-naïve first-episode psychosis patients: a randomized control trial. J Clin Psychiatry. 2006; 67: 1253-1260.
- Berg J, Stajich G, Zdanowicz. Atypical antipsychotic-induced type 2 diabetes. pharmacy times. 2012. Available at: https://www.pharmacytimes.com/publications/issue/2012/march2012/olanzapine-and-clozapine-atypical-antipsychotic-induced-type-2-diabetes-. Accessed: May 22, 2017.
- FDA Patient Safety News Show #28, June 2004. Accessed at: www.accessdata.fda.gov/psn/printer- full.cfm? id=32. Accessed November 8, 2009.
- Meyer JM. A retrospective comparison of weight, lipid, and glucose changes between risperidone- and olanzapine-treated inpatients: metabolic outcomes after 1 year. J Clin Psychiatry. 2002; 63: 425-33.
- Wu RR, Zhao JP, Liu ZN. et al. Effects of typical and atypical antipsychotics on glucose-insulin homeostasis and lipid metabolism in first-episode schizophrenia. Psychopharmacology. 2006; 186: 572-8.
- Wilton LV, Heeley EL, Pickering RM, et al. Comparative study of mortality rates and cardiac dysrhythmias in post-marketing surveillance studies of sertindole and two other atypical antipsychotic drugs, risperidone and olanzapine. J Psychopharmacology. 2001; 15: 120-6.
- Killian JG, Kerr K, Lawrence C, et al. Myocarditis and cardiomyopathy associated with clozapine. Lancet. 1999; 354: 1841-5.
- La Grenade L, Graham D, Trontell A. Myocarditis and cardiomyopathy associated with clozapine use in the United States. N Engl J Med. 2001; 345: 224-5.
- Yasui-Furukori N, Fujii A. Worsened hypertension control induced by aripiprazole. Neuropsychiatr Dis Treat. 2013; 9: 505-7.
- Khasawneh FT, Shankar GS. Minimizing cardiovascular adverse effects of atypical antipsychotic drugs in patients with schizophrenia. Cardiol Res Pract. 2014; 2014: 273060. doi: 10.1155/2014/273060.
- Sartorius N. Physical illness in people with mental disorders. World Psychiatry. 2007; 6: 3-4.
- Meyer JM. Antipsychotics and metabolics in the post-CATIE era. Curr Top Behav Neurosci. 2010; 4: 23-42.
- Rozan G, Tuchin T, Kurland M. Some implications of sexual activity for mental illness. Mental Hygiene. 1971; 55: 318–323.
- Lyketsos GC, Sakka P, Mailis A. The sexual adjustment of chronic schizophrenics: a preliminary study. British Journal of Psychiatry. 1983; 143: 376–382.
- Margolis RL, Ross CA. Expansion explosion: new clues to the pathogenesis of repeat expansion neurodegenerative diseases. Trends Mol Med. 2001; 7(11): 479-82.
- Wan H, Goodkind D, Kowal P. An Aging World: 2015. Washington, DC; 2016. Accessed at: https://census.gov/content/dam/Census/library/publications/2016/demo/p95-16-1.pdf.
- Holtzer R, Tang M-X, Devanand DP, et al. Psychopathological features in Alzheimer’s disease: course and relationship with cognitive status. J Am Geriatr Soc. 2003; 51(7):953-960.
- Factor SA, McDonald WM, Goldstein FC. The role of neurotransmitters in the development of Parkinson's disease-related psychosis. Eur J Neurol. 2017; 24(10): 1244-1254.
- Werner FM, Covenas R. Serotonergic Drugs: Agonists/Antagonists at Specific Serotonergic Subreceptors for the Treatment of Cognitive, Depressant and Psychotic Symptoms in Alzheimer's Disease. Curr Pharm Des. 2016; 22(14): 2064-71.
- Samudra N, Patel N, Womack KB, et al. Psychosis in parkinson disease: a review of etiology, phenomenology, and management. drugs aging. 2016; 33(12): 855-863.
- Chang A, Fox SH. Psychosis in parkinson's disease: epidemiology, pathophysiology, and management. drugs. 2016; (11): 1093-118. Review. Erratum in: Drugs. 2016; 76(13): 1319.
- Citrome L, Eramo A. Francois C., et al. Lack of tolerable treatment options for patients with schizophrenia. Neuropsychiatr Dis Treat. 2015; 16 (11): 3095-104.
- McGavin JK, Goa KL. Aripiprazole. CNS Drugs. November 2002; 16 (11): 779–786.