
Cerebral Microvascular Injury in Traumatic Brain Injury
Kimbra Kenney, Margalit Haber, Franck Amyot, Cora Davis, Angela Pronger, Carol Moore, Ramon Diaz-Arrastia*
Abstract
Traumatic cerebral vascular injury (TCVI) is a frequent, perhaps universal, feature after traumatic brain injury (TBI) and may be responsible for some TBI-related chronic disability. Because there are multiple pharmacologic and non-pharmacologic therapies that promote vascular health, TCVI is an attractive target for therapeutic intervention after TBI. The cerebral microvasculature (CMV) is a component of the neurovascular unit (NVU) coupling neuronal metabolism with local cerebral blood flow. The NVU participates in the pathogenesis of TBI, either directly from physical trauma or as part of the cascade of secondary injury that occurs after TBI. Pathologically, there is extensive microvascular injury in humans and experimental animals, identified with either conventional light microscopy or ultrastructural examination. It is seen in acute and chronic TBI and even described in chronic traumatic encephalopathy (CTE). Non-invasive, physiologic measures of cerebral microvascular function show dysfunction after TBI in humans and experimental animal models of TBI. These include imaging sequences Arterial Spin Labeling (ASL), Transcranial Doppler, Near InfraRed Spectroscopy (NIRS), etc. Understanding the pathophysiology of TCVI, a relatively under-studied component of TBI, has promise for developing novel TBI therapies.
Introduction
TBI places an enormous burden on patients and society, and has led to extensive preclinical research and numerous, unsuccessful clinical trials1. While the complex molecular and cellular mechanisms responsible for TBI-associated deficits are incompletely understood, substantial data suggest that traumatic cerebral vascular injury (TCVI), at least partially, underlies a significant fraction of TBI-related disability. Because the cerebral vasculature is highly plastic, TCVI is an attractive target for therapies. There are well established pharmacologic and non-pharmacologic approaches that promote vascular health, such as PDE-5 inhibitors, HMG-CoA reductase inhibitors, HDL mimetics, and PPAR-γ agonists, among others2. This review will focus on TCVI preclinical and clinical data.
The neurovascular unit
The micro-network regulating cerebral blood flow, vascular permeability and angiogenesis has been coined the neurovascular unit (NVU)3-5. It actively participates in the pathogenesis of many brain disorders, including common conditions such as hypertension, diabetes and neurodegenerative disorders (e.g. Alzheimer’s disease)5,6. The NVU has been an intense focus of research in multiple acute and chronic neurologic disorders7.
Pathophysiology of NVU injury after TBI
TCVI can result from both primary and secondary injury (e.g. blood brain barrier (BBB) disruption, increased intracellular calcium, mitochondrial dysfunction, neuroinflammation)7. After TBI, the changes seen in the BBB have a biphasic mode of action--immediate changes caused by direct damage to endothelial cells followed by changes from secondary injuries in other elements of the NVU (neurons, astrocytes, perictyes, microglia and the extracellular matrix)8. Diminished CBF and focal tissue hypoxia is a common precipitant of NVU pathophysiology and is mediated through multiple pathophysiologic cascades (e.g. BBB disruption, edema, focal ischemia)3,9,10. When injured, the NVU rapidly increases blood flow and oxygen supply and induces factors that promote angiogenesis8,11. However, the NVU also attempts self-repair through mechanisms with potential deleterious consequences that enhance secondary injury if homeostasis is not quickly restored8. These changes often occur remote from the TBI impact and represent secondary NVU changes.
Pathology of microvascular injury in acute TBI-primary injury
Preclinical studies of acute changes
Microvascular injury is a near universal finding in experimental TBI, and has been reported in nearly all animal models, including impact acceleration12,13, fluid percussion injury14, and controlled cortical impact (CCI)15. Early studies with fluid percussion injury showed peri-contusion petechial hemorrhages around small venules, pyknotic neurons, and swollen astrocytes. Ultrastructural analysis revealed early vessel wall damage in areas with irreversible neuronal injury16. Another fluid percussion injury study showed reduced microvascular density (57% loss) within cortical contusions17. After CCI, acute migration of pericytes was observed from microvascular locations to thinning areas of the basal lamina18. and, after CCI injury, acute migration of pericytes from microvascular locations to thinning areas of the basal lamina18. Electron microscopy in primates showed endothelial changes at 3 hours that persisted 1 week post-injury19. Sangiorgi et al.20 described microvascular injury changes similar to those found in humans the first 3 weeks after injury11. CMV casts taken 3 hours after CCI showed extravasation consistent with subarachnoid, subdural and intraparenchymal haemorrhage, a result of primary injury. By 12 hours, the major finding was microvascular constriction and distal caliber reduction, potentially reflecting cytotoxic edema20. A study of microvascular pathology in the CCI model at both acute and chronic time points shows microvascular injury associated with inflammation, BBB disruption and progressive white matter injury21.
Human neuropathological observations in acute/subacute TBI
TCVI is a near universal feature of severe TBI13,22. There are abundant pathological reports describing TCVI after fatal TBI23, and, although less frequently, also in individuals who died from non-TBI related complications after mild TBI11,24. Microscopic perivascular hemorrhages are seen even when macroscopic hemorrhage is absent. Microscopically, there are abundant intravascular microthrombi in the microvasculature. In samples from non-contused or contused sections after fatal TBI cases, intravascular microthrombi are seen in both, only varying in density, and correlated with focal areas of neuronal necrosis24, suggesting a possible link between microthrombi and neuronal death16. Rodriquez-Baeza and colleagues studied the CMV ultrastructurally in 10 TBI patients who died between 1 and 20 days after injury11. CMV corrosion casts revealed 3 changes in the arterioles and capillaries of the middle and deep cortical vascular zones in TBI brains: 1) longitudinal folds; 2) sunken vascular surfaces with craters at endothelial junctions; 3) reduction of the vessel lumen (Fig. 1)11.
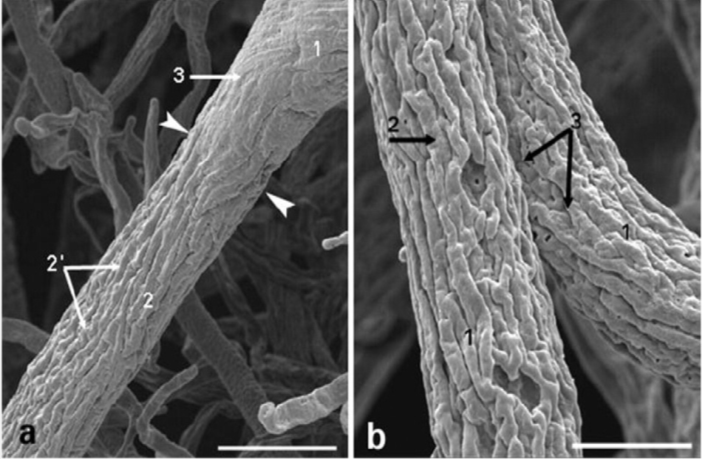
Figure 1: Scanning electron micrographs showing microvascular changes in 2 arteriolar vessels from the frontal lobe after TBI. a: Arteriole with longitudinal folds (2′) with transition from an undamaged, smooth vessel (1) to an injured, folded vessel (2) (arrowheads indicate the transition point). 1 = subpial zone; 2 = superficial zone; 3 = cortical zone. Scale bar = 86 μm. b: Arteriolar vessels (1) with longitudinal folds (2) and nuclear imprints of endothelial cells (3) at higher power. Scale bar = 23 μm.Ref: Rodriguez-Baeza A, Reina-de la Torre F, Poca A et al. Morphological features in human cortical brain microvessels after head injury: a three-dimensional and immunocytochemical study. Anat Rec A Discov Mol Cell Evol Biol 2003; 273:583–593.
Recent electron microscopy studies after severe TBI describe the following capillaries and perictyes changes: thickening of the basement membrane 3 to 8 times normal; rarefaction, vacuolization and splitting of the capillary basement membrane; pericyte hypertrophy; pericyte rarefaction and necrosis; lipofuscin and lipid deposits in pericyte cytoplasm25,26, indicating widespread microvascular injury after TBI11,25,26.
Pathology of microvascular injury in chronic TBI— CMV repair
Injured blood vessels respond to TBI through local repair (Table 1). The CMV affects this through increased protein synthesis along with cell hypertrophy and hyperplasia7. These time-dependent responses are not seen until 3 hours to 7 weeks after TBI and can be used in forensic injury dating22. In animal models, therapies that promote CMV repair and/or angiogenesis, such as statins27 or sildenafil28,29, promote neurologic recovery.
Pathology of cerebral microvascular injury in chronic traumatic encephalopathy (CTE)
Prominent microvasculopathy is also described in CTE30,31. Cortical vascular changes, such as thickened perforating arteries, absence of nuclei in vascular cells, and diffuse hyaline, were described in the initial CTE cases32. Among CTE cases in the Corsellis collection diffuse perivascular hemosiderin collections in macrophages, neuroglia or extracellular space were described33. In sports-related CTE cases, striking vascular changes such as perivascular microgliosis and astrocytosis, neurofibrillary tangles, and spindle-shaped neurites in the sulcal depths of cortical gyri, and hemosiderin-laden perivascular macrophages have been described34. In a series of blast-associated CTE cases, McKee similarly noted perivascular lymphocytic cuffing, and hemosiderin-laden macrophages within cerebral vessel walls and focal calcifications of penetrating small thalamic and deep white matter vessels35 similar to that seen in mouse models of TCVI22,30.
Non-invasive assessment of TCVI
There are two methods to assess CMV function: functional neuroimaging and cerebrovascular reactivity (CVR) measurements. Novel neuroimaging sequences have been coupled with dynamic procedures that increase physiologic demand to measure the CMV’s ability to respond and indirectly TCVI.
Neuroimaging and CVR in experimental animals
Neuroimaging studies consistently show reduced cerebral blood flow (CBF) acutely after experimental TBI by functional MRI36and laser Doppler flowometry37. In fluid percussion injury, CBF measured via continuous ASL, showed reductions the first 2 weeks, corresponding with decreases in cortical small vessel density36.
CVR can be assessed in experimental models through cranial windows that allow direct visualization of the pial microvasculature. The anesthetized animals undergo a hypercapnia challenge with 3-5% carbon dioxide (CO2) while the pial microvasculature is assessed. One week after injury, there is a significant decrease in CVR compared to sham injured controls12. Other studies report that TBI causes a loss of the normal vasodilatory response to potent vasodilators (acetylcholine, adenosine, and sodium nitroprusside)14.
Neuroimaging studies in humans
CBF has been extensively studied after TBI in humans, especially in the acute period, when CBF deficits are common38. Bonne et al.38 used single photon emission computed tomography (SPECT) to measure regional CBF in symptomatic, chronic TBI patients and found areas of cerebral hypoperfusion. With SPECT, Lewine et al.39 described CBF abnormalities in 40% of 30 chronic symptomatic mild TBI patients and found SPECT to be significantly more sensitive than MRI. A recent meta-review concluded that SPECT outperformed CT and MRI in both acute and chronic TBI diagnosis40. In all 10 studies that compared SPECT to CT or MRI, SPECT identified CBF deficits that were not seen by conventional imaging.
Other advanced MRI techniques have been helpful in evaluating TCVI. ASL provides a direct measurement of arterial perfusion in absolute units of CBF. Kim et al. showed that chronic moderate or severe TBI patients have reduced global CBF in the resting state, as well as decreased regional perfusion in the thalamus, posterior cingulate cortex, and frontal cortex41. Regional CBF can also be calculated by perfusion-weighted imaging (PWI). In 15 symptomatic sports-related concussion patients studied 6 months post-concussion, PWI showed reduced CBF in the thalami bilaterally and reduced cerebral blood volume in the left thalamus compared to controls9. Susceptibility-weighted imaging (SWI) detects microbleeds better than gradient recalled echo MRI in traumatic axonal injury, and the total number and volume of microbleeds correlate with TBI-associated functional outcomes42. Traumatic microbleeds are seen in 23% of mild TBI patients scanned between 8 and 60 days after injury, and their presence inversely correlates with neurocognitive testing43.
Assessment of cerebrovascular reactivity in humans
Several methods exist to study CVR non-invasively in humans after hypercapnia, breath-holding, or acetazolamide44, using Transcranial Doppler (TCD), functional MRI and near infra-red spectroscopy (NIRS). While TCD and NIRS offer the advantage of high temporal resolution, MRI offers superb spatial resolution. A prospective study of 299 moderate-to-severe TBI patients assessed cerebral vasospasm with TCD45. Nearly half (45.2%) of the patients had vasospasm with the highest risk at day 3 after injury. CVR, measured by both TCD and NIRS, was decreased in 12 professional boxers46 72 hours after a bout. Compared to controls the boxers also had chronically impaired CVR by both modalities and lower CVR measurements correlated with increased neurocognitive dysfunction and inversely correlated with TBI exposure. A meta-analysis reported reduced CVR via TCD in 42 athletes examined between 2 and 5 days after sports-related concussion47.
NIRS, another noninvasive measure of CVR, is currently also being used to assess CMV function in TBI. NIRS allows CVR measurements during dynamic challenges that are independent of hemoglobin concentration, skull thickness and extracranial circulation48. Using a NIRS-based CVR index in 40 acute TBI patients, total hemoglobin reactivity index measured by NIRS correlated with the intracranial pressure derived cerebrovascular pressure reactivity index49. A study of 37 critically ill TBI patients showed a good correlation between hemoglobin reactivity index measured via NIRS and intracranial pressure.
Hypercapnia-BOLD MRI reliably measures deficits in CVR in the chronic stage after TBI
Direct measures of microvascular injury require assessment of CVR using manipulations that directly affect endothelial function. One method combines MRI with the Blood Oxygen Dependent (BOLD) signal in response to hypercapnia challenge50. The hypercapnia challenge is administered during the MRI by the inhalation of room air alternating with room air + 5% CO2. The CVR is the ratio of change in BOLD signal to the change in end-tidal CO2 and with voxel-by-voxel measurements, a CVR map can be drawn (Fig. 2,3).
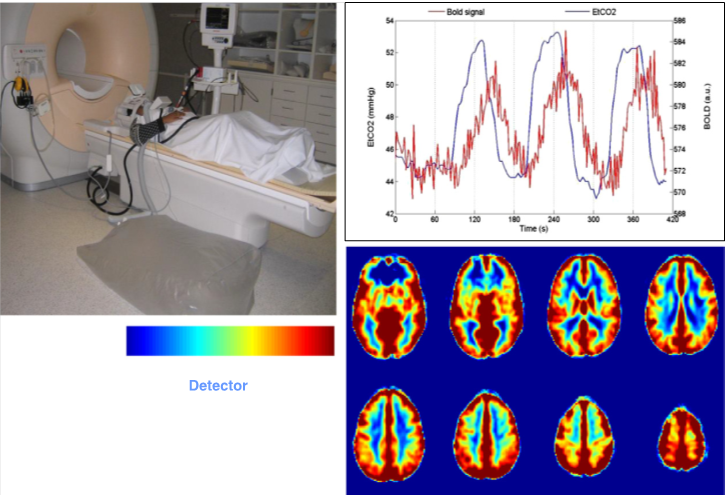
Figure 2: Top left: MRI-BOLD with 5% CO2 hypercapnia challenge set-up with patient shown on the MRI stretcher and Douglas bag with 5% CO2 at the side on the floor. Top right: Typical tracing of end tidal CO2 in blue and MRI-BOLD signal superimposed during MRI-BOLD with hypercapnia challenge. Bottom right: Typical CVR map generated with MRI-BOLD with hypercapnia challenge after coregistration with T1 MRI image. Ref: Kenney K, Amyot F, Haber M, et al. Cerebral Vascular Injury in Traumatic Brain Injury. Exp Neurol. 2016;275 Pt 3:353-366. Courtesy of Franck Amyot, Ph.D., CNRM, USUHS.
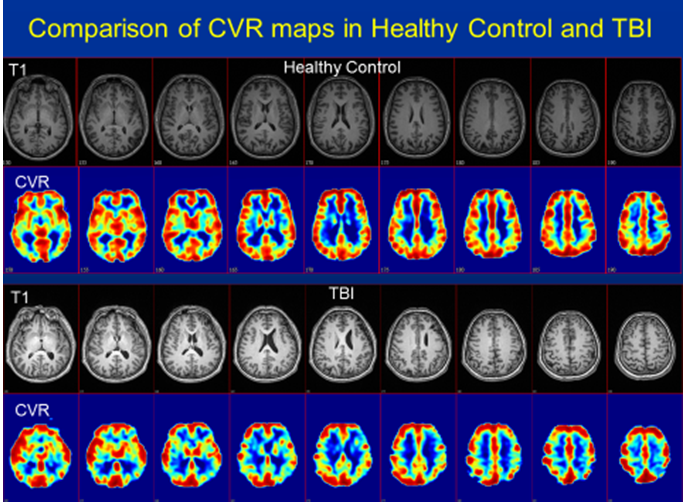
Figure 3: T1 MRI images (rows 1 & 3) with corresponding cerebrovascular reactivity (CVR) maps (rows 2 & 4) based on MRI-BOLD with 5% hypercapnia challenge in a healthy control (top 2 rows) compared to chronic TBI subject (bottom 2 rows). Note the relatively uniform CVR throughout the cortex in the control subject in the top two rows compared to the patchy, moth-eaten appearance to the CVR maps from the TBI subjects. As expected, in areas of visible encephalomalacia, the CVR was decreased. However, there were also many regions where the CVR was depressed but the structural MRI was normal. Ref: Courtesy of Franck Amyot, Ph.D., CNRM, USUHS.
Our group recently completed a study of CVR by MRI-BOLD with hypercapnia challenge in 27 chronic stage TBI patients (15 controls). There was significantly decreased CVR in the chronic TBI subjects (Fig. 4)4. We adapted the hypercapnia challenge to NIRS testing and found similar CVR reductions in chronic TBI, and a strong correlation with the MRI-BOLD results (Fig. 5). These results support the persistence of TCVI into the chronic stage and the reliable use of both NIRS and MRI. CVR may prove to be a useful predictive and pharmacodynamic biomarker in therapeutic trials of TCVI.
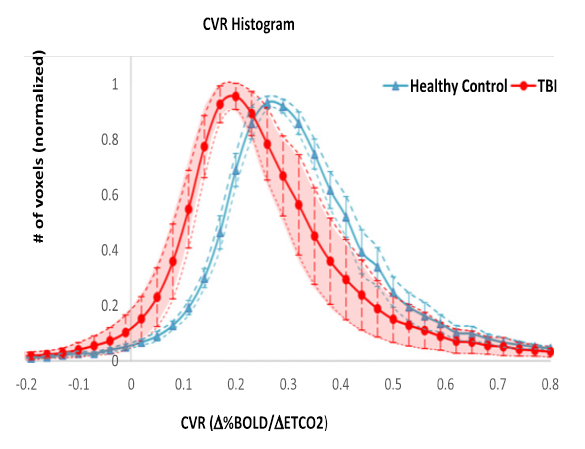
Figure 4: Cerebrovascular reactivity (CVR) histogram via MRI-BOLD with 5% CO2 hypercapnia challenge in moderate and severe traumatic brain injury patients (red) compared to age-matched healthy controls (blue). The CVR histogram in TBI patients is both shifted to the left and has much greater variance. The CVR histogram in healthy controls is remarkably stable with very little intersubject variability. Ref: Kenney K, Amyot F, Haber M, et al. Cerebral Vascular Injury in Traumatic Brain Injury. Exp Neurol. 2016;275 Pt 3:353-366. Courtesy of Franck Amyot, Ph.D., CNRM, USUHS.
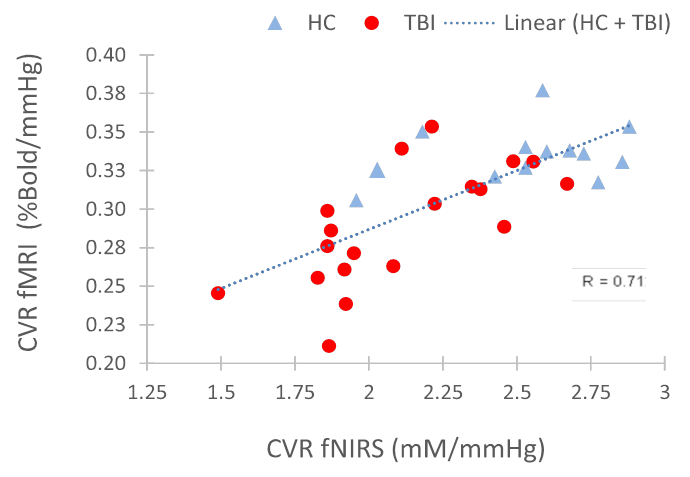
Figure 5: Correlation of CVR measurements via MRI-BOLD (with 5% CO2 hypercapnia challenge via the Douglas Bag method) and fNIRS (Near InfraRed Spectroscopy with 5% CO2 hypercapnia challenge via the Douglas Bag method) in traumatic brain injury (TBI) and healthy controls. The complementary methods give similar results with a high degree of correlation (R = 0.71) with lower and more variable CVR measurements in TBI patients compared to healthy controls. Ref: Kenney K, Amyot F, Haber M, et al. Cerebral Vascular Injury in Traumatic Brain Injury. Exp Neurol. 2016;275 Pt 3:353-366. Courtesy of Franck Amyot, Ph.D., CNRM, USUHS.
Conclusions
TCVI has been an under-recognized TBI phenotype despite a robust theoretical construct and a large body of empirical evidence. The studies reviewed here support the hypothesis that TCVI is near ubiquitous after TBI. Further, it plays a potentially important role in chronic post-concussive symptoms and even TBI-associated neurodegenerative disorders. Further neuropathological studies are required to characterize the extent and time course of TCVI. As novel vascular technologies are developed, we will achieve a better understanding of TCVI’s role in clinical symptoms both acutely and remotely after TBI. Understanding the pathophysiology of TCVI will aid in developing effective treatments targeting the underlying pathology.
Acknowledgements
Work in the authors’ laboratory was supported by the Center for Neuroscience and Regenerative Medicine, and by the Chronic Effects of Neurotrauma Consortium. The contents of this paper are solely the responsibility of the authors and do not represent the official views of the Department of Defense or the Center for Neuroscience and Regenerative Medicine.
References
- Narayan RK, Michel ME, Ansell B, et al. Clinical trials in head injury. J Neurotrauma. 2002;19(5):503-557.
- Nicolakakis N, Hamel E. Neurovascular function in Alzheimer's disease patients and experimental models. J Cereb Blood Flow Metab. 2011;31(6):1354-1370.
- Lecrux C, Hamel E. The neurovascular unit in brain function and disease. Acta Physiol (Oxf). 2011;203(1):47-59.
- Kenney K, Amyot F, Haber M, et al. Cerebral Vascular Injury in Traumatic Brain Injury. Exp Neurol. 2016;275 Pt 3:353-366.
- Zlokovic BV. Neurovascular pathways to neurodegeneration in Alzheimer's disease and other disorders. Nat Rev Neurosci. 2011;12(12):723-738.
- Zlokovic BV. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57(2):178-201.
- Golding EM. Sequelae following traumatic brain injury. The cerebrovascular perspective. Brain Res Brain Res Rev. 2002;38(3):377-388.
- Shlosberg D, Benifla M, Kaufer D, Friedman A. Blood-brain barrier breakdown as a therapeutic target in traumatic brain injury. Nat Rev Neurol. 2010;6(7):393-403.
- Bartnik-Olson BL, Holshouser B, Wang H, et al. Impaired neurovascular unit function contributes to persistent symptoms after concussion: a pilot study. J Neurotrauma. 2014;31(17):1497-1506.
- Xing C, Hayakawa K, Lok J, Arai K, Lo EH. Injury and repair in the neurovascular unit. Neurol Res. 2012;34(4):325-330.
- Rodriguez-Baeza A, Reina-de la Torre F, Poca A, Marti M, Garnacho A. Morphological features in human cortical brain microvessels after head injury: a three-dimensional and immunocytochemical study. Anat Rec A Discov Mol Cell Evol Biol. 2003;273(1):583-593.
- Baranova AI, Wei EP, Ueda Y, Sholley MM, Kontos HA, Povlishock JT. Cerebral vascular responsiveness after experimental traumatic brain injury: the beneficial effects of delayed hypothermia combined with superoxide dismutase administration. J Neurosurg. 2008;109(3):502-509.
- Gao G, Oda Y, Wei EP, Povlishock JT. The adverse pial arteriolar and axonal consequences of traumatic brain injury complicated by hypoxia and their therapeutic modulation with hypothermia in rat. J Cereb Blood Flow Metab. 2010;30(3):628-637.
- Wei EP, Hamm RJ, Baranova AI, Povlishock JT. The long-term microvascular and behavioral consequences of experimental traumatic brain injury after hypothermic intervention. J Neurotrauma. 2009;26(4):527-537.
- Cherian L, Goodman JC, Robertson C. Improved cerebrovascular function and reduced histological damage with darbepoietin alfa administration after cortical impact injury in rats. J Pharmacol Exp Ther. 2011;337(2):451-456.
- Dietrich WD, Alonso O, Halley M. Early microvascular and neuronal consequences of traumatic brain injury: a light and electron microscopic study in rats. J Neurotrauma. 1994;11(3):289-301.
- Corsellis JA, Brierley JB. Observations on the pathology of insidious dementia following head injury. J Ment Sci. 1959;105:714-720.
- Dore-Duffy P, Owen C, Balabanov R, Murphy S, Beaumont T, Rafols JA. Pericyte migration from the vascular wall in response to traumatic brain injury. Microvasc Res. 2000;60(1):55-69.
- Maxwell RJ, Martinez-Perez I, Cerdan S, et al. Pattern recognition analysis of 1H NMR spectra from perchloric acid extracts of human brain tumor biopsies. Magn Reson Med. 1998;39(6):869-877.
- Sangiorgi S, De Benedictis A, Protasoni M, et al. Early-stage microvascular alterations of a new model of controlled cortical traumatic brain injury: 3D morphological analysis using scanning electron microscopy and corrosion casting. J Neurosurg. 2013;118(4):763-774.
- Glushakova OY, Johnson D, Hayes RL. Delayed increases in microvascular pathology after experimental traumatic brain injury are associated with prolonged inflammation, blood-brain barrier disruption, and progressive white matter damage. J Neurotrauma. 2014;31(13):1180-1193.
- Hausmann R, Betz P. The time course of the vascular response to human brain injury--an immunohistochemical study. Int J Legal Med. 2000;113(5):288-292.
- Tomlinson BE. Brain-stem lesions after head injury. J Clin Pathol Suppl (R Coll Pathol). 1970;4:154-165.
- Stein SC, Graham DI, Chen XH, Smith DH. Association between intravascular microthrombosis and cerebral ischemia in traumatic brain injury. Neurosurgery. 2004;54(3):687-691; discussion 691.
- Castejon OJ. Ultrastructural pathology of cortical capillary pericytes in human traumatic brain oedema. Folia Neuropathol. 2011;49(3):162-173.
- Castejon OJ. Ultrastructural alterations of human cortical capillary basement membrane in human brain oedema. Folia Neuropathol. 2014;52(1):10-21.
- Wu H, Jiang H, Lu D, et al. Induction of angiogenesis and modulation of vascular endothelial growth factor receptor-2 by simvastatin after traumatic brain injury. Neurosurgery. 2011;68(5):1363-1371; discussion 1371.
- Zhang R, Wang Y, Zhang L, et al. Sildenafil (Viagra) induces neurogenesis and promotes functional recovery after stroke in rats. Stroke. 2002;33(11):2675-2680.
- Pifarre P, Prado J, Giralt M, Molinero A, Hidalgo J, Garcia A. Cyclic GMP phosphodiesterase inhibition alters the glial inflammatory response, reduces oxidative stress and cell death and increases angiogenesis following focal brain injury. J Neurochem. 2010;112(3):807-817.
- Goldstein LE, Fisher AM, Tagge CA, et al. Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci Transl Med. 2012;4(134):134ra160.
- McKee AC, Stern RA, Nowinski CJ, et al. The spectrum of disease in chronic traumatic encephalopathy. Brain. 2013;136(Pt 1):43-64.
- Corsellis JA, Bruton CJ, Freeman-Browne D. The aftermath of boxing. Psychol Med. 1973;3(3):270-303.
- Adams CW, Bruton CJ. The cerebral vasculature in dementia pugilistica. J Neurol Neurosurg Psychiatry. 1989;52(5):600-604.
- McKee AC, Daneshvar DH, Alvarez VE, Stein TD. The neuropathology of sport. Acta Neuropathol. 2014;127(1):29-51.
- McKee AC, Robinson ME. Military-related traumatic brain injury and neurodegeneration. Alzheimers Dement. 2014;10(3 Suppl):S242-253.
- Hayward NM, Tuunanen PI, Immonen R, Ndode-Ekane XE, Pitkanen A, Grohn O. Magnetic resonance imaging of regional hemodynamic and cerebrovascular recovery after lateral fluid-percussion brain injury in rats. J Cereb Blood Flow Metab. 2011;31(1):166-177.
- Thomale UW, Kroppenstedt SN, Beyer TF, Schaser KD, Unterberg AW, Stover JF. Temporal profile of cortical perfusion and microcirculation after controlled cortical impact injury in rats. J Neurotrauma. 2002;19(4):403-413.
- Bonne O, Gilboa A, Louzoun Y, et al. Cerebral blood flow in chronic symptomatic mild traumatic brain injury. Psychiatry Res. 2003;124(3):141-152.
- Lewine JD, Davis JT, Bigler ED, et al. Objective documentation of traumatic brain injury subsequent to mild head trauma: multimodal brain imaging with MEG, SPECT, and MRI. J Head Trauma Rehabil. 2007;22(3):141-155.
- Raji CA, Tarzwell R, Pavel D, et al. Clinical utility of SPECT neuroimaging in the diagnosis and treatment of traumatic brain injury: a systematic review. PLoS One. 2014;9(3):e91088.
- Kim J, Whyte J, Patel S, et al. Resting cerebral blood flow alterations in chronic traumatic brain injury: an arterial spin labeling perfusion FMRI study. J Neurotrauma. 2010;27(8):1399-1411.
- Tong KA, Ashwal S, Holshouser BA, et al. Diffuse axonal injury in children: clinical correlation with hemorrhagic lesions. Ann Neurol. 2004;56(1):36-50.
- Huang YL, Kuo YS, Tseng YC, Chen DY, Chiu WT, Chen CJ. Susceptibility-weighted MRI in mild traumatic brain injury. Neurology. 2015;84(6):580-585.
- Kassner A, Roberts TP. Beyond perfusion: cerebral vascular reactivity and assessment of microvascular permeability. Top Magn Reson Imaging. 2004;15(1):58-65.
- Oertel M, Boscardin WJ, Obrist WD, et al. Posttraumatic vasospasm: the epidemiology, severity, and time course of an underestimated phenomenon: a prospective study performed in 299 patients. J Neurosurg. 2005;103(5):812-824.
- Bailey DM, Jones DW, Sinnott A, et al. Impaired cerebral haemodynamic function associated with chronic traumatic brain injury in professional boxers. Clin Sci (Lond). 2013;124(3):177-189.
- Gardner AJ, Tan CO, Ainslie PN, et al. Cerebrovascular reactivity assessed by transcranial Doppler ultrasound in sport-related concussion: a systematic review. Br J Sports Med. 2015;49(16):1050-1055.
- Kainerstorfer JM, Sassaroli A, Hallacoglu B, Pierro ML, Fantini S. Practical steps for applying a new dynamic model to near-infrared spectroscopy measurements of hemodynamic oscillations and transient changes: implications for cerebrovascular and functional brain studies. Acad Radiol. 2014;21(2):185-196.
- Zweifel C, Castellani G, Czosnyka M, et al. Noninvasive monitoring of cerebrovascular reactivity with near infrared spectroscopy in head-injured patients. J Neurotrauma. 2010;27(11):1951-1958.
- Lu H, Liu P, Yezhuvath U, Cheng Y, Marshall O, Ge Y. MRI mapping of cerebrovascular reactivity via gas inhalation challenges. J Vis Exp. 2014(94).