
DNA-PK Deficiency in Alzheimer's Disease
Jyotshna Kanungo*
Abstract
Alzheimer’s disease (AD) is characterized by neuronal death with an accumulation of intra-cellular neurofibrillary tangles (NFT) and extracellular amyloid plaques. Reduced DNA repair ability has been reported in AD brains. In neurons, the predominant mechanism to repair double-strand DNA breaks (DSB) is non-homologous end joining (NHEJ) that requires DNA-dependent protein kinase (DNA-PK) activity. DNA-PK is a holoenzyme comprising the p460 kD DNA-PK catalytic subunit (DNA-PKcs) and its activator Ku, a heterodimer of p86 (Ku80) and p70 (Ku70) subunits. Upon binding to double-stranded DNA ends, Ku recruits DNA-PKcs to process NHEJ. In AD brains, reduced NHEJ activity as well as DNA-PKcs and Ku protein levels have been shown. Normal aging brains also show a reduction in both DNA-PKcs and Ku levels questioning a direct link between NHEJ ability and AD, and suggesting additional players/events in AD pathogenesis. Deficiency of Ku80, a somatostatin receptor, can disrupt somatostatin signaling thus inducing amyloid beta (Aβ) generation, which in turn can potentiate DNA-PKcs degradation and consequently loss of NHEJ activity, an additional step negatively affecting DSB repair. Trigger of these two different pathways culminating in genome instability may differentiate the outcomes between AD and normal aging.
Alzheimer’s disease (AD) is a CNS neurodegenerative disease, characterized by specific neuronal death with accumulated neurofibrillary tangles (NFT) and extracellular amyloid beta (Aβ) deposits1. Aβ directly injures neocortical and limbic system neuron2. It also indirectly activates the microglia that release pro-inflammatory cytokines and reactive oxygen species (ROS), both events being neurotoxic3,4. Other factors linked to the development of AD include apolipoprotein E genotype5; hyperphosphorylation of cytoskeletal proteins (neurofilaments and Tau)6, and Aβ metabolism7. As diverse as the pathological and biochemical presentations of AD are8, no single factor has been confirmed as the sole cause of this complex disease9-12. Studies have shown a link between oxidative stress (e.g., ROS) and AD pathogenesis11,13,14. Since oxidative stress can cause DNA lesions, changes in the levels and activity of DNA-repair proteins have garnered special interest of study of AD patients or patients with mild cognitive impairment15,16.
Cellular damage by oxidative stress caused by the generation of ROS has been implicated in pathophysiology of AD as well as normal aging and elevated levels of oxidative damage in DNA, both nuclear and mitochondrial, have been observed in AD brains17. As DNA damage accumulates and DNA repair process lags or goes awry, a potentially adverse scenario can set in contributing to AD18,19. Some human hereditary genetic defects in the DNA repair system also manifest in early onset of developmental and progressive neurodegeneration20,21. Cells use several types of DNA repair systems such as base excision repair (BER), nucleotide excision repair (NER), single strand break repair (SSBR), and double strand break repair (DSBR). Of all these various DNA damages, double strand break (DSB) happens to be the most lethal. There are two major DSB repair pathways in the eukaryotes; non-homologous end joining (NHEJ) and homologous recombination (HR). NHEJ, the predominant pathway for DSBR in higher order organisms, functions throughout the cell cycle22,24, whereas HR functions are confined to the S and G2 stages of the cell cycle25. DNA-PK plays an essential role in accessing the DNA ends during NHEJ26,27.
As a response to DNA damage, expression and activity of many kinases including members of the PI3 kinase family are altered28. One of these kinases, the DNA-dependent protein kinase (DNA-PK) preferentially phosphorylates the serines (S) and threonines (T) of its targets although it can also phosphorylate other S-T/hydrophobic residues29. DNA-PK holoenzyme consists of a catalytic subunit (DNA-PKcs), p460 and a regulatory subunit (Ku). The Ku protein is a heterodimer composed of 70 kD (Ku70) and 80 kD (Ku80) subunits; and possesses the ability to bind to DNA ends30,31. DNA-PK is conserved across species32,33 and participates in transcription, DNA recombination and repai34-38. In the absence of DNA-PKcs, Ku binds DNA ends in a sequence-independent manner39, however, Ku is required for targeting DNA-PKcs to damaged DNA ends in physiologic conditions in vitro and in living cells40. DSB can activate DNA-PK both in trans (occurs via kinase autophosphrylation) or cis (occurs via specific DNA strand orientation and sequence bias) modes41-43.
Post-mitotic neurons are mature, do not proliferate44,45 and are also one of the most metabolically and transcriptionally active cells (review46). Therefore, these neurons are more susceptible to suffer from risks involving DNA damage. NHEJ, unlike HR, is error-prone since it acts at the DNA break points and the repair process can cause loss of one or more nucleotides. However, since most of the higher eukaryote genome is non-coding, errors occurred during DSBR by NHEJ rarely translate into any deleterious effects. Unfortunately, as people age, accumulation of these non-obvious errors eventually can lead to genome instability, thereby causing cellular death or dysfunction. For example, 10% of p53 mutations in human cancers could be attributed to deletions arising from NHEJ sites47. NHEJ being the predominant form of DSBR pathway in post-mitotic neurons48, mouse neurons deficient in components of NHEJ, such as XLF, DNA Ligase IV, XRCC4, Ku70 and Ku80 (Fig. 1), undergo excessive apoptosis49,50. Mice with defective NHEJ show accelerated aging51,52. Loss of NHEJ activity in the developing brain causes prenatal lethality and can lead to neurodegenerative diseases in adults49,53,54.
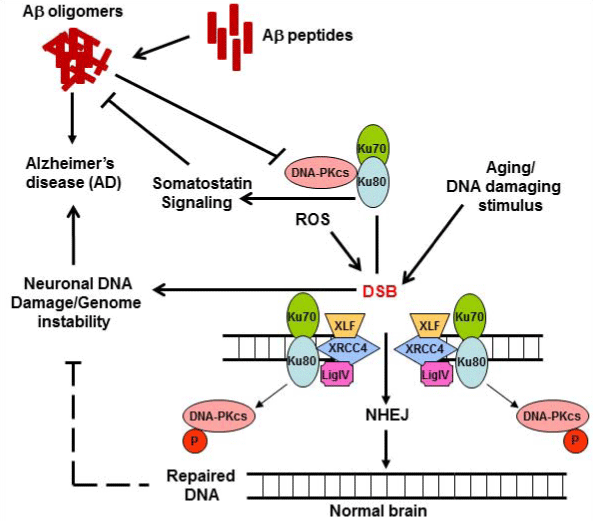
Figure 1: Schematic presentation of a potential link of DSB, DNA-PK and Aβ in AD brains.
Upon induction of DSB either by normal aging/ROS or other DNA damaging agents, Ku80/Ku70 and DNA-PKcs are rapidly recruited to DNA ends, and DNA repair occurs as it would in normal cases. However, in AD brains, in addition to formation of Aβ oligomers from Aβ peptides, sustained DSB in the genome would cause genome instability leading to the loss of normal neuronal activity. Additionally, with depleted Ku80, a somatostatin receptor, disruption of somatostatin signaling could potentially induce Aβ generation thus accelerating AD pathology. DSB: DNA double strand break; DNA-PK: DNA-dependent protein kinase; ROS: Reactive oxygen species; Aβ Amyloid beta
Terminally differentiated post-mitotic neurons triggered to re-enter cell cycle following stimuli associated with DNA damage and oxidative stress undergo apoptosis55,56. Neuronal DNA damage is linked to neurons re-entering cell cycle56,57. To this end, DNA replication may be a consequence of cell cycle re-entry preceding neurodegeneration in AD brains58. Moreover, reactive oxygen/nitrogen species reportedly cause deregulated and inefficient DNA replication known as ‘replication stress’59. It is possible that “replication stress” in AD pathogenesis can lead to genomic instability potentially resulting in Aβ accumulation and deregulated cell cycles60. Adding to this scenario, existence of defective DNA repair systems in post-mitotic neurons would lead to accumulation of further DNA damages and genomic instabilities61,62 (Fig. 1). It has been suggested that accumulated single-stranded DNA (ssDNA) at replication forks may give rise to aberrant DNA structures resulting in DSBs that activate DNA-PK63. With this scenario, in AD, reduced DNA-PK as such would further enhance DSB accumulation. Intracellular increase in DNA content observed in AD brains58,64 may also result from these combined events. Indeed, it has been reported that DNA-PKcs mutant cells under stress fail to arrest replication65. Thus, neurons deficient in DNA-PK activity could uninterruptedly undergo replication stress ending with genome instability (Fig. 1).
DNA-PK plays a critical role, first, by sensing DNA damage and then, inducing signaling pathways including programmed cell death51. Ku80-/- mice are defective in NHEJ, telomere maintenance and show premature aging52,66. Ku80 and DNA-PKcs protein levels as well as Ku80’s DNA-binding ability are reduced following severe ischemic injury leading to neuronal death in rabbit67. Furthermore, although not significantly different from the age-matched controls, Ku-DNA binding is reduced in extracts of post-mortem AD mid-frontal cortex that may be linked to reduced levels of Ku and DNA-PKcs proteins68. Reduced NHEJ activity in extracts of the cortices of AD brains compared to the normal subjects and significantly lower levels of DNA-PKcs in the AD brain extracts have also been reported69. Since DNA-PK is a critical player in cell survival/death and gene transcription, it is tempting to directly link reduced levels of DNA-PK subunits to less proficient NHEJ in AD brains and neurodegeneration. It is likely that DNA damage (e.g., induced by ROS) in neurons that are already challenged with reduced NHEJ activity, may trigger them to re-enter cell cycle albeit unsuccessfully, resulting in accumulation of excessive genomic damage leading to neuronal death. Therefore, reduced levels of DNA-PKcs and Ku80/Ku70 subunits in post-mortem AD brains may be an important upstream event that predisposes the neurons to AD.
In NGF-differentiated PC12 cells, sub-lethal levels of aggregated Aβ25-35 have been shown to inhibit DNA-PK activity as does hydrogen peroxide70. One of its potential mechanisms may be Aβ-induced ROS-mediated DNA-PKcs degradation via carbonylation, an irreversible oxidative protein modification71,72. A decrease in DNA-PKcs expression in neurons and astrocytes of AD brains73, although not significant compared to age-matched controls, has been reported74. Whether Aβ-induced attenuation of DNA-PK activity and reduced NHEJ activity (Fig. 1) leading to neurotoxicity is linked to the development of AD awaits careful scrutiny.
Ku80 has been shown to be a specific receptor for somatostatin75 and can regulate DNA-PK activity through somatostatin signaling pathways76. Somatostatin modulates both motor activity and cognition77. Somatostatinergic neurons exist in the CNS including the cerebral cortex, hippocampus, hypothalamus, and spinal cord78. Along with various other neuropeptides, somatostatin levels are significantly reduced in AD brains79 and cerebrospinal fluid80. Somatostatin receptors are also reduced in the cortical areas of the AD brain81. Loss of somatostatinergic neurons along with reduction in somatostatin transcripts in a transgenic mouse model of AD, and somatostatin deficiency potentially triggering Aβ generation have been reported82. It is possible that Ku80 deficiency can negatively affect somatostatin signaling leading to Aβ generation, thereby contributing to AD pathogenesis, a process independent of the involvement of DSB (Fig. 1).
DNA-PKcs, Ku80 and Ku70 are exceptionally abundant proteins in human cells83. Reduced level of DNA-PKcs in AD brains has been attributed to Aβ-induced proteasome-mediated degradation of DNA-PKcs71,72. Whether disruption of the somatostatin signaling due to Ku80 deficiency inducing Aβ generation precedes DNA-PKcs degradation is not known. If true, it would highlight Ku80 as a dual player in AD pathogenesis; when deficient, by indirectly promoting Aβ generation and directly causing NHEJ deficiency.
Abbreviations
AD: Alzheimer’s disease; ATM: Ataxia telangiectasia mutated protein; BER: base excision repair; DNA-PK: DNA-dependent protein kinase; DSB: double strand breaks; DSBR: Double strand break repair; HR: Homologous recombination; NER: Nucleotide excision repair; NGF: Nerve growth factor; Lig IV: Ligase IV; NHEJ: Non-homologous end-joining; NFT: Neurofibrillary tangle; SSBR: Single strand break repair; XLF: XRCC4-like factor; XRCC4: X-ray repair cross-complementing protein 4
Acknowledgements
This document has been reviewed in accordance with United States Food and Drug Administration (FDA) policy and approved for publication. Approval does not signify that the contents necessarily reflect the position or opinions of the FDA nor does mention of trade names or commercial products constitute endorsement or recommendation for use. The findings and conclusions in this report are those of the author and do not necessarily represent the views of the FDA.
References
- Smith MA, Perry G. The pathogenesis of Alzheimer disease: an alternative to the amyloid hypothesis. J Neuropathol Exp Neurol. 1997;56(2):217.
- Mucke L, Selkoe DJ. Neurotoxicity of amyloid β-protein: synaptic and network dysfunction. Cold Spring Harb Perspect Med. 2012;2(7):a006338.
- Czirr E, Wyss-coray T. The immunology of neurodegeneration. J Clin Invest. 2012;122(4):1156-63.
- Wyss-coray T, Rogers J. Inflammation in Alzheimer disease-a brief review of the basic science and clinical literature. Cold Spring Harb Perspect Med. 2012;2(1):a006346.
- Corder EH, Saunders AM, Strittmatter WJ, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science. 1993;261(5123):921-3.
- Trojanowski JQ, Schmidt ML, Shin RW, Bramblett GT, Rao D, Lee VM. Altered tau and neurofilament proteins in neuro-degenerative diseases: diagnostic implications for Alzheimer's disease and Lewy body dementias. Brain Pathol. 1993;3(1):45-54.
- Selkoe DJ. Alzheimer's disease: genotypes, phenotypes, and treatments. Science. 1997;275(5300):630-1.
- Scheltens P, Blennow K, Breteler MM, de Strooper B, Frisoni GB, Salloway S et al. Alzheimer's disease. Lancet. 2016; doi: 10.1016/S0140-6736(15)01124-1
- Barnes J, Dickerson BC, Frost C, Jiskoot LC, Wolk D, Van der flier WM. Alzheimer's disease first symptoms are age dependent: Evidence from the NACC dataset. Alzheimers Dement. 2015;11(11):1349-57.
- Crutch SJ, Schott JM, Rabinovici GD, Boeve BF, Cappa SF, Dickerson BC, et al. Shining a light on posterior cortical atrophy. Alzheimers Dement. 2013; 9:463-5.
- Murray IV, Proza JF, Sohrabji F, Lawler JM. Vascular and metabolic dysfunction in Alzheimer's disease: a review. Exp Biol Med (Maywood). 2011;236(7):772-82.
- Van der flier WM, Pijnenburg YA, Fox NC, Scheltens P. Early-onset versus late-onset Alzheimer's disease: the case of the missing APOE ?4 allele. Lancet Neurol. 2011;10(3):280-8.
- Aldred S, Bennett S, Mecocci P. Increased low-density lipoprotein oxidation, but not total plasma protein oxidation, in Alzheimer's disease. Clin Biochem. 2010; 43:267-71.
- Bennett S, Grant MM, Aldred S. Oxidative stress in vascular dementia and Alzheimer's disease: a common pathology. J Alzheimers Dis. 2009;17(2):245-57.
- Bennett G, Papamichos-chronakis M, Peterson CL. DNA repair choice defines a common pathway for recruitment of chromatin regulators. Nat Commun. 2013;4:2084.
- Bucholtz N, Demuth I. DNA-repair in mild cognitive impairment and Alzheimer's disease. DNA Repair (Amst). 2013;12(10):811-6.
- Wang J, Xiong S, Xie C, Markesbery WR, Lovell MA. Increased oxidative damage in nuclear and mitochondrial DNA in Alzheimer's disease. J Neurochem. 2005;93(4):953-62.
- Adamec E, Vonsattel JP, Nixon RA. DNA strand breaks in Alzheimer's disease. Brain Res. 1999;849(1-2):67-77.
- Love S, Barber R, Wilcock GK. Apoptosis and expression of DNA repair proteins in ischaemic brain injury in man. Neuroreport. 1998;9(6):955-9.
- Gueven N, Chen P, Nakamura J, et al. A subgroup of spinocerebellar ataxias defective in DNA damage responses. Neuroscience. 2007;145(4):1418-25.
- Lee Y, Mckinnon PJ. Responding to DNA double strand breaks in the nervous system. Neuroscience. 2007;145(4):1365-74.
- Critchlow SE, Jackson SP. DNA end-joining: from yeast to man. Trends Biochem Sci. 1998;23(10):394-8.
- Lieber MR. The biochemistry and biological significance of nonhomologous DNA end joining: an essential repair process in multicellular eukaryotes. Genes Cells. 1999;4(2):77-85.
- Pastink A, Eeken JC, Lohman PH. Genomic integrity and the repair of double-strand DNA breaks. Mutat Res. 2001;480-481:37-50.
- Rothkamm K, Krüger I, Thompson LH, Löbrich M. Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol Cell Biol. 2003;23(16):5706-15.
- Kienker LJ, Shin EK, Meek K. Both V(D)J recombination and radioresistance require DNA-PK kinase activity, though minimal levels suffice for V(D)J recombination. Nucleic Acids Res. 2000;28(14):2752-61.
- Oksenych V, Kumar V, Liu X, et al. Functional redundancy between the XLF and DNA-PKcs DNA repair factors in V(D)J recombination and nonhomologous DNA end joining. Proc Natl Acad Sci USA. 2013;110(6):2234-9.
- Bensimon A, Aebersold R, Shiloh Y. Beyond ATM: the protein kinase landscape of the DNA damage response. FEBS Lett. 2011;585(11):1625-39.
- Lees-Miller SP, Meek K. Repair of DNA double strand breaks by non-homologous end joining. Biochimie. 2003; 85:1161-73.
- De vries E, Van driel W, Bergsma WG, Arnberg AC, Van der vliet PC. HeLa nuclear protein recognizing DNA termini and translocating on DNA forming a regular DNA-multimeric protein complex. J Mol Biol. 1989;208(1):65-78.
- Mimori , Hardin JA. Mechanism of interaction between Ku protein and DNA. J Biol Chem. 1986; 261:10375-9.
- Kanungo J. Prolonged incubation in seawater induces a DNA-dependent protein phosphorylation activity in Arbacia punctulata eggs. Biochem Biophys Res Commun. 2002;294(3):667-71.
- Kanungo J. DNase I-resistant DNA-dependent protein kinase activity in Xenopus oocytes. Mol Cell Biochem. 2008;309(1-2):33-40.
- Carter T, Vancurová I, Sun I, Lou W, Deleon S. A DNA-activated protein kinase from HeLa cell nuclei. Mol Cell Biol. 1990;10(12):6460-71.
- Jackson SP, Macdonald JJ, Lees-miller S, Tjian R. GC box binding induces phosphorylation of Sp1 by a DNA-dependent protein kinase. Cell. 1990;63(1):155-65.
- Satoh MS, Lindahl T. Role of poly(ADP-ribose) formation in DNA repair. Nature. 1992;356(6367):356-8.
- Liang F, Romanienko PJ, Weaver DT, Jeggo PA, Jasin M. Chromosomal double-strand break repair in Ku80-deficient cells. Proc Natl Acad Sci USA. 1996;93(17):8929-33.
- Kanungo J, Wang HY, Malbon CC. Ku80 is required but not sufficient for Galpha13-mediated endodermal differentiation in P19 embryonic carcinoma cells. Biochem Biophys Res Commun. 2004; 323:293-8.
- Walker JR, Corpina RA, Goldberg J. Structure of the Ku heterodimer bound to DNA and its implications for double-strand break repair. Nature. 2001;412(6847):607-14.
- Drouet J, Delteil C, Lefrançois J, Concannon P, Salles B, Calsou P. DNA-dependent protein kinase and XRCC4-DNA ligase IV mobilization in the cell in response to DNA double strand breaks. J Biol Chem. 2005;280(8):7060-9.
- Dobbs TA, Tainer JA, Lees-miller SP. A structural model for regulation of NHEJ by DNA-PKcs autophosphorylation. DNA Repair (Amst). 2010;9(12):1307-14.
- Nagasawa H, Little JB, Lin YF, et al. Differential role of DNA-PKcs phosphorylations and kinase activity in radiosensitivity and chromosomal instability. Radiat Res. 2011;175(1):83-9.
- Reddy YV, Ding Q, Lees-miller SP, Meek K, Ramsden DA. Non-homologous end joining requires that the DNA-PK complex undergo an autophosphorylation-dependent rearrangement at DNA ends. J Biol Chem. 2004;279(38):39408-13.
- Korr H. Proliferation of different cell types in the brain of senile mice autoradiographic studies with 3H- and 14C-thymidine. Exp Brain Res. 1982;Suppl 5:51-7.
- Ridet JL, Malhotra SK, Privat A, Gage FH. Reactive astrocytes: cellular and molecular cues to biological function. Trends Neurosci. 1997;20(12):570-7.
- Rao KS . DNA repair in aging rat neurons. Neuroscience. 2007; 145:1330-40.
- Greenblatt MS, Grollman AP, Harris CC. Deletions and insertions in the p53 tumor suppressor gene in human cancers: confirmation of the DNA polymerase slippage/misalignment model. Cancer Res. 1996;56(9):2130-6.
- Rass U, Ahel I, West SC. Defective DNA repair and neurodegenerative disease. Cell. 2007;130(6):991-1004. 10.1016/j.cell.2007.08.043
- Brooks PJ. DNA repair in neural cells: basic science and clinical implications. Mutat Res. 2002;509(1-2):93-108.
- Sekiguchi JM, Gao Y, Gu Y, et al. Nonhomologous end-joining proteins are required for V(D)J recombination, normal growth, and neurogenesis. Cold Spring Harb Symp Quant Biol. 1999;64:169-81.
- Smith GC, Jackson SP.The DNA-dependent protein kinase. Genes Dev. 1999; 13:916-34.
- Vogel H, Lim DS, Karsenty G, Finegold M, Hasty P. Deletion of Ku86 causes early onset of senescence in mice. Proc Natl Acad Sci USA. 1999;96(19):10770-5.
- Mckinnon PJ, Caldecott KW. DNA strand break repair and human genetic disease. Annu Rev Genomics Hum Genet. 2007;8:37-55.
- Yang Y, Herrup K. Loss of neuronal cell cycle control in ataxia-telangiectasia: a unified disease mechanism. J Neurosci. 2005;25(10):2522-9.
- Krantic S, Mechawar N, Reix S, Quirion R. Molecular basis of programmed cell death involved in neurodegeneration. Trends Neurosci. 2005;28(12):670-6.
- Kruman II, Wersto RP, Cardozo-pelaez F, et al. Cell cycle activation linked to neuronal cell death initiated by DNA damage. Neuron. 2004;41(4):549-61.
- McMurray CT. To die or not to die: DNA repair in neurons. Mutat Res. 2005; 577:260-74.
- Yang Y, Geldmacher DS, Herrup K. DNA replication precedes neuronal cell death in Alzheimer's disease. J Neurosci. 2001;21(8):2661-8.
- Shen C, Lancaster CS, Shi B, Guo H, Thimmaiah P, Bjornsti MA. TOR signaling is a determinant of cell survival in response to DNA damage. Mol Cell Biol. 2007;27(20):7007-17.
- Yurov YB, Vorsanova SG, Iourov IY. The DNA replication stress hypothesis of Alzheimer's disease. ScientificWorldJournal. 2011;11:2602-12.
- Bester AC, Roniger M, Oren YS, et al. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell. 2011;145(3):435-46.
- Burhans WC, Weinberger M. DNA replication stress, genome instability and aging. Nucleic Acids Res. 2007;35(22):7545-56.
- Buisson R, Boisvert JL, Benes CH, Zou L. Distinct but Concerted Roles of ATR, DNA-PK, and Chk1 in Countering Replication Stress during S Phase. Mol Cell. 2015;59(6):1011-24.
- Chen J, Cohen ML, Lerner AJ, Yang Y, Herrup K. DNA damage and cell cycle events implicate cerebellar dentate nucleus neurons as targets of Alzheimer's disease. Mol Neurodegener. 2010;5:60.
- Liu S, Opiyo SO, Manthey K, et al. Distinct roles for DNA-PK, ATM and ATR in RPA phosphorylation and checkpoint activation in response to replication stress. Nucleic Acids Res. 2012;40(21):10780-94.
- Pandita TK. The role of ATM in telomere structure and function. Radiat Res. 2001;156(5 Pt 2):642-7.
- Shackelford DA, Tobaru T, Zhang S, Zivin JA. Changes in expression of the DNA repair protein complex DNA-dependent protein kinase after ischemia and reperfusion. J Neurosci. 1999;19(12):4727-38.
- Shackelford DA. DNA end joining activity is reduced in Alzheimer's disease. Neurobiol Aging. 2006;27(4):596-605.
- Davydov V, Hansen LA, Shackelford DA. Is DNA repair compromised in Alzheimer's disease?. Neurobiol Aging. 2003;24(7):953-68.
- Cardinale A, Racaniello M, Saladini S, et al. Sublethal doses of β-amyloid peptide abrogate DNA-dependent protein kinase activity. J Biol Chem. 2012;287(4):2618-31.
- Grune T, Reinheckel T, Davies KJ. Degradation of oxidized proteins in mammalian cells. FASEB J. 1997;11(7):526-34.
- Nyström T. Role of oxidative carbonylation in protein quality control and senescence. EMBO J. 2005;24(7):1311-7.
- Kanungo J. DNA-dependent protein kinase and DNA repair: relevance to Alzheimer's disease. Alzheimers Res Ther. 2013;5(2):13.
- Simpson JE, Ince PG, Haynes LJ, et al. Population variation in oxidative stress and astrocyte DNA damage in relation to Alzheimer-type pathology in the ageing brain. Neuropathol Appl Neurobiol. 2010;36(1):25-40.
- Le romancer M, Reyl-desmars F, Cherifi Y, et al. The 86-kDa subunit of autoantigen Ku is a somatostatin receptor regulating protein phosphatase-2A activity. J Biol Chem. 1994;269(26):17464-8.
- Pucci S, Bonanno E, Pichiorri F, Mazzarelli P, Spagnoli LG. The expression and the nuclear activity of the caretaker gene ku86 are modulated by somatostatin. Eur J Histochem. 2004;48(2):103-10.
- Gillies G. Somatostatin: the neuroendocrine story. Trends Pharmacol Sci. 1997;18(3):87-95.
- Johansson O, Hökfelt T, Elde RP. Immunohistochemical distribution of somatostatin-like immunoreactivity in the central nervous system of the adult rat. Neuroscience. 1984;13(2):265-339.
- Chi LM, Wang X, Nan GX. In silico analyses for molecular genetic mechanism and candidate genes in patients with Alzheimer's disease. Acta Neurol Belg. 2016.
- Burgos-Ramos E, Hervas-Aguilar A, Aguado-Llera D, Puebla-Jimenez L, Hernandez-Pinto AM, Barrios V et al. Somatostatin and Alzheimer's disease. Mol Cell Endocrinol. 2008; 286:104-11.
- Beal MF, Mazurek MF, Tran VT, Chattha G, Bird ED, Martin JB. Reduced numbers of somatostatin receptors in the cerebral cortex in Alzheimer's disease. Science. 1985;229(4710):289-91.
- Saiz-sanchez D, Ubeda-bañon I, De la rosa-prieto C, et al. Somatostatin, tau, and beta-amyloid within the anterior olfactory nucleus in Alzheimer disease. Exp Neurol. 2010;223(2):347-50.
- Mimori T, Hardin JA, Steitz JA. Characterization of the DNA-binding protein antigen Ku recognized by autoantibodies from patients with rheumatic disorders. J Biol Chem. 1986;261(5):2274-8.