
Imbalances of Tripartite Synapses Responsible for the Pathophysiology of Mental Disorders and Epilepsy
Bernhard J. Mitterauer*
Volitronics-Institute for basic research and psychopathology, Salzburg, Austria
Abstract
A model of imbalances in tripartite synapses responsible for the pathophysiology of mental disorders and epilepsy is reviewed. A tripartite synapse consists of the presynapse and the postsynapse as the neuronal component and the astrocyte as the glial component. Based on a formalism of system-balancing it is hypothesized that the expression of astroglial receptors determines imbalances of neurotransmission. In depression, tripartite synapses are imbalanced since neurotransmitters cannot activate the overexpressed astroglial receptors in the time leading to a prolonged neurotransmission. Inversely, in mania the imbalance of tripartite synapses is caused by a surplus of neurotransmitters overactivating underexpressed astroglial receptors causing a shortened neurotransmission. If astroglial receptors are non-functional, they cannot be activated by neurotransmitters leading to an unconstrained neurotransmission responsible for schizophrenia. In epilepsy, astroglial receptors are overexpressed, but glutamatergic synapses are hyperactivated and GABAergic synapses are hypoactivated causing an imbalance between excitatory and inhibitory tripartite synapses responsible for epileptogenesis. It is suggested that common imbalances of astroglia-synapse interactions may be responsible for mental disorders and epilepsy.
Introduction
Synaptic dysregulations or imbalances are commonly found in neuropsychiatric disorders and epilepsy. Recently, it is becoming increasingly clear that dysregulations and structural changes of astroglia play a central role in the pathophysiology of these disorders1-3. Here, I will focus on imbalances in tripartite synapses mainly determined by the expression of astroglial receptors. A tripartite synapse consists of the presynapse and the postsynapse as the neuronal component and the astrocyte as the glial component4.
The structure and basic functions of tripartite synapses is well established5-7. Elementary mechanisms of balancing have been identified in both the neuronal synapses and tripartite synapses where astrocyte coordinate synaptic networks generating a balanced excitation and inhibition8,9. This review outlines my models of imbalanced tripartite synapses that may be responsible for the pathophysiology of depression, mania, schizophrenia and, epilepsy.
Logic of System-Balancing
The operations between astroglial receptors (acr) and neurotransmitters (NT) can be described by a formalism of system-balancing10,11,12,13. Figure 1 shows four elementary system states in tripartite synapses deduced from the formalism applied. If the concentration of NT, formally interpreted as values, is appropriate for the occupancy of acr, interpreted as variables, the synaptic state is in balance (a). In the case of an overexpression of acr, the amount of NT is not sufficient for the complete occupancy of the acr leading to an imbalanced synaptic system. This system state operates in depression (b). If acr are underexpressed, a surplus of NT arises. This system state is imbalanced operating in mania (c). In the case of non-functional acr they cannot be activated by NT representing a totally unbalanced synaptic system state responsible for the pathophysiology of schizophrenia (d).
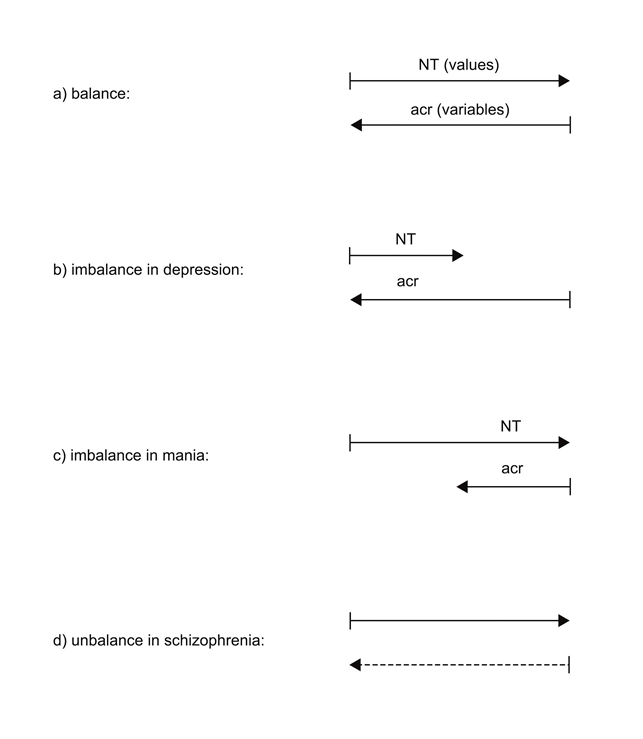
Figure 1: System-balancing in tripartite synapses
Neurotransmitter (NT) represent values, astroglial receptors (acr) variables signified by two inverse parallel lines. If NT (values) correspond to acr (variables), the synaptic system is in balance (a). If acr dominate NT (shorter upper line), the synaptic system is imbalanced causing depression (b). If acr are underexpressed (shorter lower line) in relation to the concentration of NT, the synaptic system is imbalanced causing mania (c). If acr are non-functional (dashed line), the system is totally unbalanced responsible for schizophrenia (d). (modified11)
Imbalanced Tripartite Synapses in Depression
Figure 2 outlines a model of an underbalanced tripartite synapse responsible for the pathophysiology of depression14,15. The upregulation of gap junctions (GJ, red) exerts an overexpression of acr that cannot be activated by NT in real time leading to a diminished Ca2+ concentration (↓) and an underproduction of gliotransmitters (GT) (↓). Consequently,the negative feedback on presynaptic receptors (psr) is protracted. Since acr outnumber the available NT, a relative lack of NT arises and neurotransmission is prolonged. This may explain the main symptoms of depression such as psychomotor retardation, feelings of insufficiency and disturbed circadian rhythms.
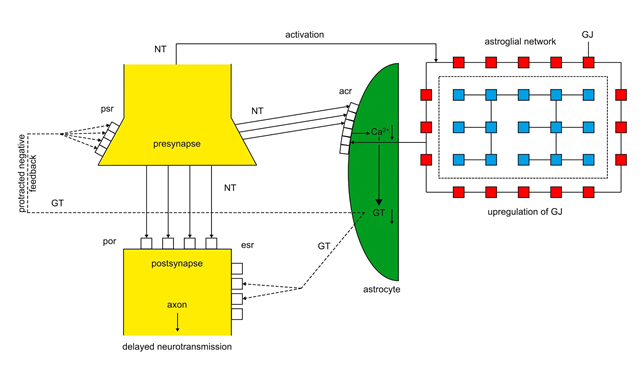
Figure 2: Imbalanced tripartite synapse in depression
Neurotransmitters (NT) released from the presynapse activate postsynaptic receptors (por) and astroglial receptors (acr). Overexpressed gap junctions (GJ, red) upregulate the expression of acr. Overexpressed acr cannot be completely activated by NT causing diminished Ca2+ concentration (↓) and production of gliotransmitters (GT, ↓). This leads to a protracted activation of por, extrasynaptic receptors (esr) and a protracted negative feedback on presynaptic receptors (psr) delaying neurotransmission. (dashed lines)15
There is some evidence that acr are upregulated in depression. Adenosine A2A receptors are overexpressed in animal models of chronic stress and polymorphisms of A2A receptors are associated with emotional disturbances or depression16. Moreover, overexpressed acr have been identified in Parkinson’s disease17, Alzheimer’s disease18, and temporal lobe epilepsy3.
Imbalanced Tripartite Synapses in Mania
The imbalance of astroglia-synapse interactions in mania occurs, if gap junctions in the astroglial network and acr are underexpressed. This leads to a surplus of NT relative to the underexpressed acr causing a flooding of acr with NT. As shown in figure 3 the overactivation of acr increases the Ca2+ concentration and GT production that exert a shortened feedback on the psr accelerating neurotransmission. This may explain the main symptoms of mania as manic distractibility, the flight of ideas, overactivity and circadian-biorhythmic disturbances such as insomnia19,20.
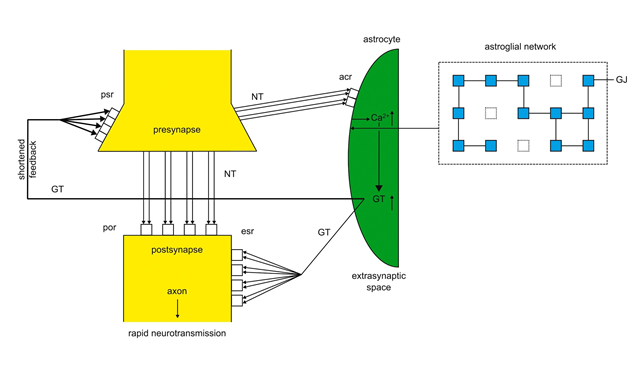
Figure 3: Imbalanced tripartite synapse in mania
Underexpression of gap junctions (dashed squares) downregulates the expression of acr. Underexpressed acr are flooded (bold lines) by NT increasing Ca2+ concentration (↑) and the production of GT(↑) leading to a shortened feedback on psr (bold line). por and esr are also hyperactivated causing a rapid neurotransmission. (modified13)
Although excessive neurotransmission caused by the underexpression of acr is as yet not identified, relevant studies indicate excessive dopaminergic activity in brains with mania or bipolar disorder21.
Unbalanced Tripartite Synapses in Schizophrenia
The model of schizophrenia is based on unbalanced astroglia-synapse interactions caused by non-functional acr leading to an unconstrained synaptic flux so that astroglia loses their modulatory function in tripartite synapses22,23. As shown in figure 4 non-functional acr (crosses) cannot be activated by NT. Hence the production of GT is not possible and GT cannot exert a negative feedback on cognate psr leading to an unconstrained neurotransmission. Given that patients with schizophrenia are unable to distinguish between the self and the others, the model proposed is explanatory. Since the flux of neurotransmission cannot be modulated by astroglia, a gap between the sensory information processing of the neuronal system and the “inner” astroglial network exists resulting in incoherent thoughts, delusions, and affective flattening23.
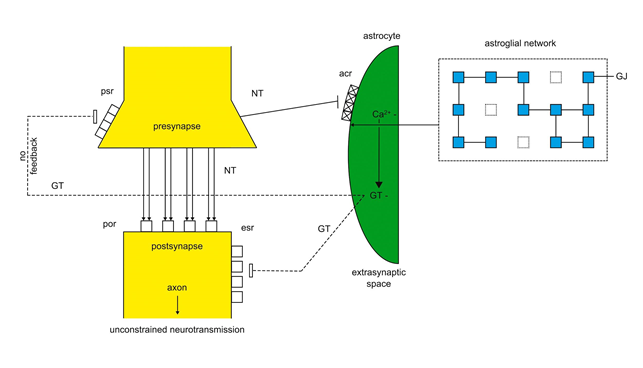
Figure 4: Unbalanced tripartite synapses in schizophrenia
acr are non-functional (crosses) and cannot be activated by NT. Ca2+ and GT production is not activated by NT leading to an unconstrained neurotransmission (bold arrows), since no negative feedback is exerted by NT on psr (dashed line).
Recent experimental findings indicate that alterations in excitatory signaling, particularly involving hypofunction of N-methyl-D-aspartate receptors (NMDR) play a key role in the schizophrenia disease process24. Theoretically, a model of the pathophysiology of schizophrenia focusing on non-functional gliotransmission in tripartite synapse has been mathematically described25.
Imbalanced Tripartite Synapses in Epilepsy
In addition to the various recognized findings in the neuronal system, there is growing evidence that dysfunctional astrocytes are crucial players in epilepsy3,26,27. Here, I outline a model of imbalanced tripartite synapses responsible for the pathophysiology of epilepsy based on the imbalance between hyperactivated glutamatergic and hypoactivated GABAergic tripartite synapses.
Figure 5a depicts a hyperactivated glutamatergic tripartite synapse. Glutamate (GLU) is excessively released from the hyperactivated presynapse flooding both postsynaptic receptors (por) and acr. As identified in temporal lobe epilepsy metabotropic GLU receptors (mGluR) in astrocytes are overexpressed and become overactivated by GLU. This hyperactivation of mGluR causes excessive Ca2+ concentrations and GT production in the astrocyte. GTs hyperactivate psr and esr exerting a shortened feedback and an excessive depolarization of the postsynapse. Note, overexpressed mGluR are not balanced by GLU, but hyperactivated because of the flooding with GLU. The hyperactivated astrocyte may exert excitotoxic effects leading to a decoupling of gap junctions in the astroglial network3.
In parallel, inverse mechanisms may operate in hypoactivated GABAergic tripartite synapses shown in figure 5b. Here, acr are also overexpressed, but the production of GABA is decreased and unable to completely activate acr. Consequently, Ca2+ concentration in astroglia is too low and the underproduction of GT exerts a protracted negative feedback on psr. Therefore, GABAergic tripartite synapses cannot put into action their inhibitory effect in time causing an imbalance (bold,double-headed arrow) between hyperactivated and hypoactivated tripartite synapses. This imbalance may play a key role in epileptogenesis28.
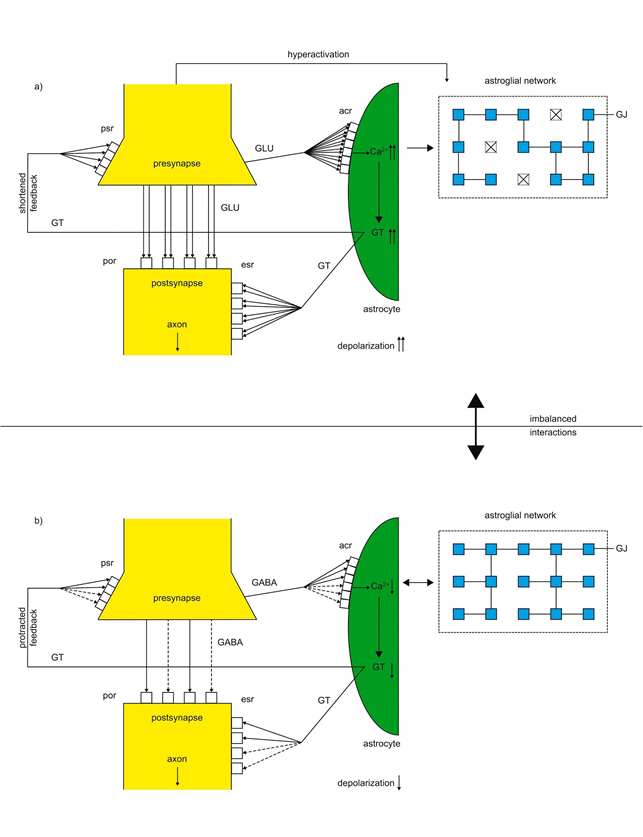
Figure 5: Imbalance between glutamatergic and GABAergic tripartite synapses in epilepsy
a.) Glutamate (GLU) hyperactivates por and acr (bold lines). The Ca2+ concentration and GT production is excessively increased (↑↑) leading to a shortened feedback on psr and the hyperactivation of esr (bold lines) excessively depolarizes the postsynapse.
b.) Decreased GABA release (dashed lines) from the presynapse hypoactivates por and acr. Ca2+ and GT production is diminished (↓) causing a protracted feedback on psr and a protracted depolarization of the postsynapse, since esr are hypoactivated by GT (dashed lines). The interactions between the glutamatergic and GABAergic tripartite synapses are imbalanced (bold double-headed arrow).(modified13)
Concluding Remarks
In the perspective of common pathophysiological mechanisms operating in mental disorders and epilepsy, the determining function of acr is comparable in these disorders. Considering epilepsy, on the one hand, acr are overexpressed in glutamatergic tripartite synapses and flooded by GLU comparable to imbalanced synapses in mania. On the other hand, acr are also overexpressed in GABAergic tripartite synapses but hypoactivated by GABA comparable to imbalanced synapses in depression. The model here proposed is mainly theoretical and must be clinically and biologically tested. Concerning depression, we could show in a computer simulation that prolonged synaptic information processing may be responsible for retarded depressive behavior29. This result is clinically supported by a sample of patients with major depressive disorder30. In addition, comprehensive case studies on depression and schizophrenia indicate that the model of synaptic imbalance based on astroglia-synapse interactions may be explanatory for the main symptoms of these disorders14,31.
Admittedly, the testing of the abnormal expression of acr in animal models and in patients with neuropsychiatric disorders with imaging techniques has not yet been conducted, but should be done in the near future.
Acknowledgements
I am indebted to Christian Streili for designing the figures and tables, and Marie Motil for preparing the final version of the paper.
References
- Chung WS, Welsh CA, Barres BA, et al. Do glia drive synaptic and cognitive impairment in disease? Nat Neurosci. 2015; 18: 1539-1545.
- Verkhratsky A, Rodrigues JJ, Steardo L. Astrogliopathy: a central element of neuropsychiatric diseases. Neuroscientist. 2014; 20: 576-588.
- Binder DK, Steinhäuser C. Role of astrocyte dysfunction in epilepsy. Neurosci and Biobehav Psychol. 2017. http://dx.doi.org/10.016/B978-0-12-809324-5-00071-7
- Arague A, Parpura V, Sanzgiri RP, et al. Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci. 1999; 22: 208-2015.
- Perea G, Navarrete M, Arague A. Tripartite synapses: astrocytes process and control synaptic information. Trends Neurosci. 2009; 32: 421-431.
- DePitta M, Brunel N, Volterra A. Astrocytes: orchestrating synaptic plasticity. Neurosci. 2016; 323: 43-61.
- Pereira A, Furlan FA. Astrocytes and human cognition: modeling information integration and modulation of neuronal activity. Prog Neurobiol. 2010; 92: 405-420.
- Fellin T, Pascual O, Hayden PG. Astrocytes coordinate synaptic networks: balanced excitation and inhibition. Physiology. 2006; 21: 208.2015.
- Robertson JM. Astrocyte domains and seamless expression of consciousness and explicit memories. MEHY. 2013; 81; 1017-1024.
- Guenther G. Das Bewusstsein der Maschinen. Agis Verlag, Krefeld; 1963.
- Mitterauer BJ. Imbalance of glial-neuronal interaction in synapses: a possible mechanism of the pathophysiology of bipolar disorder. Neuroscientist. 2004; 10: 199.206.
- Mitterauer BJ. Synaptic imbalances in endogenous psychoses. Biosystems. 2010; 100: 113-121.
- Mitterauer BJ. Balancing and imbalancing of astrocytic receptors in tripartite synapses. Common pathophysiological model of mental disorders and epilepsy. MEHY. 2015; 84: 315.320.
- Mitterauer BJ, Narziss, Echo. Ein psychobiologisches Modell der Depression. Springer Vienna. 2009.
- Mitterauer BJ. Towards a comprehensive psychobiological model of major depressive disorder. Open Journal of Depression. 2018; 7: 31-49.
- Coelho JE, Alves P, Canas PM, et al. Overexpression of adenosine A2A receptors in rats: effects on depression, locomotion, and anxiety. Front Psychiatry. 2014; 5: 67.doi: 10.3389/fpsyt.2014.00067
- Ishida Y, Nagai A, Kobayashi S, et al. Upregulation of protease-activated receptor 1 in astrocytes in Parkinson’s disease: astrocyte-mediated neuroprotection through levels of glutathione peroxidase.
- Yu W, Mechavar N, Kratic S et al. Upregulation of astrocytic alpha 7 nicotinic receptors in Alzheimer’s disease brain- possibly relevant to amyloid pathology. Mol Neurodegen. 7 (Suppl 1) : 07. http://www.molecularneurodegeneration.com/content/7/S1/07
- American Psychiatric Association. Diagnostic and statistical manual of mental disorders (5th ed.), Washington, DC; 2013.
- Mitterauer BJ. Pseudoomnipotence: a model of the manic syndrome. In: Kotlar M (ed.), Nova Science Publishers, New York, 161-178.
- Berk M, Dodd S, Kauer- Sant’Anna M, et al. Dopamine dysregulation syndrome: Implications for a dopamine hypothesis of bipolar disorder. Acta Psychiatr Scand Suppl. 2007; 116: 41-910.
- Mitterauer BJ. Pathophysiology of schizophrenia based on impaired glial-neuronal interactions. OJMP. 2014; 3: 126-140.
- Mitterauer BJ, Kofler-Westergren B. Possible effects of synaptic imbalances on oligodendrocyte-axonic interactions in schizophrenia. Front Psychiat. 2011. doi.10.3389/fpsyt 2011 00015.
- Balu DT. The NMDA receptor and schizophrenia: from pathophysiology to treatment. Adv Pharmacol. 2016; 76: 351-382.
- Mahdavi A, Bahrami F, Janahmad M. Analysis of impaired LTP in schizophrenia using an extended mathematical model of a tripartite synapse. 22nd Iranian Conference on Biomedical Engineering, CBME. 2015.
- Coulter DA, Steinhäuser C. Role of astrocytes in epilepsy. Cold Spring Harb Perspect Med. 2015; 25 (3): a022434. Doi. 10.1101/cshperspect.a022434
- Losi G, Cammarota M, Carmignoto G. The role of astroglia in the epileptic brain. Front Pharmacol. 2012. doi:10.3389/fphar201200132.
- Robel S, Sontheimer H. Glia as drivers of abnormal neuronal activity. Nat Neurosci. 2016; 19: 28-33.
- Mitterauer BJ. Excess of astrocyte receptors in tripartite synapses may cause a severe disorder of information processing and behavior in depression. The Bi-Monthly Journal of the BWW Society. 2008; Vol. VIII, No 3.
- Rothuber H, Mitterauer B. Comprehensive behavioral analysis of patients with major depressive disorder. Med Sci Monit. 2011; 17: CR1-CR7.
- Mitterauer B. Verlust der Selbst Grenzen. Entwurf einer interdisziplinären Theorie der Schizophrenie. Springer Vienna. 2005.