
Kv1.3 In Microglia: Neuroinflammatory Determinant and Promising Pharmaceutical Target
Sara R. Roig1, Irene Estadella1, Sergi Cirera-Rocosa1, María Navarro-Pérez1, Antonio Felipe1*
1Molecular Physiology Laboratory, Dpt. de Bioquímica i Biomedicina Molecular, Institut de Biomedicina (IBUB), Universitat de Barcelona, Av. Diagonal 643, E-08028 Barcelona, Spain
Abstract
Microglial cells are responsible for brain immunosurveillance. These cells maintain brain homeostasis, phagocytose cellular debris, present antigens to T-lymphocytes and secrete cytokines and chemokines. Upon activation, microglia can polarize into two different phenotypes in particular environments: the classical M1 proinflammatory state and the alternative M2 anti-inflammatory state. Chronically activated M1 microglial cells are involved in neuroinflammation in various brain diseases. In multiple sclerosis, Alzheimer’s disease, Parkinson’s disease and neuropathic pain, brain inflammation is involved in initiation and pathological progression, which is partially mediated by activated microglia. Molecular studies revealed that expression of the voltage-gated potassium channel Kv1.3 is a M1 phenotypic feature. Indeed, an increase in Kv1.3 expression and function was detected in chronically activated microglial cells in patients. Therefore, recent works evaluated Kv1.3-targeted therapies for neuroimmune diseases and showed encouraging results. Inhibition of Kv1.3 activity led to amelioration of some pathology-related features in brain diseases. This review summarizes the latest findings concerning microglial function in neuroinflammatory processes, focusing on the involvement of Kv1.3 and highlighting this channel as a promising therapeutic target.
Microglia in Neuroinflammatory Diseases
Microglial cells are the main innate immune effector cells of the brain. Constantly scrutinizing the environment, these cells detect and react to injury and insults (immunosurveillance). Upon activation, microglia phagocytoses cellular debris, present antigens to T-cells and produce the cytokines and chemokines governing an intricate pluricellular response1. Microglia also plays key roles in homeostasis by regulating cell death and neurogenesis engulfing synaptic material2. Likewise, synaptic maturation links to microglial function3. The acute inflammatory response is advantageous and necessary for resolving a transient pathophysiological situation. However, chronic inflammation can lead to functional alteration and tissue damage. Nervous system inflammation (called neuroinflammation) can be particularly deleterious, contributing to pathogenesis in the peripheral and central nervous systems2. Indeed, low-level sustained neuroinflammation accompanied by a redox imbalance might cause and drive the disease progression4.
Alzheimer’s disease (AD) is the most prevalent neurodegenerative chronic disease. The main pathological features of this disorder include extracellular senile plaques formed by amyloid-β peptide (Aβ) and intracellular aggregates of the microtubule-associated and phosphorylated protein tau2. In addition to these neuronal events, AD progression and even initiation also involves the immune system5. Amyloid precursor protein (APP) activates microglia6. Indeed, AD brains exhibited activated microglia, which endocytoses pathogenic Aβ peptides while producing survival-promoting signals. However, prolonged inflammation generates synaptotoxic cytokines, chemokines and reactive oxygen/nitrogen species7. Further studies in the human brain showed Aβ plaques surrounded by IL-1β-positive microglia8. Moreover, impairment of microglial activation in a murine AD model exerted protective effects on hippocampal neurogenesis and improved cognitive defects9.
Parkinson’s disease (PD) is characterized by the progressive death of dopaminergic neurons in the substantia nigra pars compacta and the accumulation of Lewy bodies enriched in α-synuclein protein. Similar to the situation in AD, neuroinflammation participates in the exacerbation of the illness2. Dopamine neurons are highly sensitive to inflammatory injury, suggesting a role for microglia in PD progression5. Evidence indicates that pathological α-synuclein facilitates the progression of PD via the activation of the transcription factor NF-ιB, which further increases the expression of proinflammatory cytokines, such as TNF-α and IL-1β10. Moreover, mutations in leucine-rich repeat kinase 2 compromise the ability of microglial cells to internalize and degrade α-synuclein, thus triggering neuroinflammation11.
During the progression of multiple sclerosis (MS), the involvement of microglia seems minor, although these cells are recruited to MS plaques. In this context, mast cells play major roles during the neuroinflammatory MS progression2. However, activation of microglia and astrocytes is a characteristic of amyotrophic lateral sclerosis (ALS)12. The crosstalk between mast cells and microglia further activates both cell types. Thus, microglia becomes more sensitive to damage-associated molecular patterns (i.e., high mobility group box 1), which are elevated in the spinal cord of ALS patients13. The association between mast cells and neuromuscular junction (NMJ) denervation indicates that after pathogenic events, mast cells infiltrate and degranulate at NMJs, which correlates with progressive NMJ degradation2.
The main cause of disability in the developed world is stroke, and 8 of 10 cases are due to cerebral ischemia2. The stroke-associated pathology suggests activation of microglia, release of pro-inflammatory cytokines/chemokines and infiltration of neutrophils/macrophages14. Interestingly, microglia, together with astrocytes and oligodendrocytes are pivotal for conditioning the microenvironment after traumatic brain injury (TBI)15,16. Moreover, a long-term investigation in humans showed chronically activated microglia years after the traumatic event17. Similarly, studies in rodents have shown upregulated proinflammatory markers, chronically activated microglia, injury extension and myelin damage18. Therefore, the therapeutic inhibition of microglial activation has been proposed as beneficial2.
Treatments attenuating peripheral neuropathic pain (NP) mainly focus on reducing pain transduction19. However, the origin and maintenance of this pathological condition depend mainly on astrocytes, microglia and Schwann cells, together with systemic immune cells20. Activated spinal microglia produces IL-1β to modulate neuronal activity via either membrane receptors or proinflammatory signals. Indeed, the expression of P2X and P2Y purinergic receptors in dorsal horn microglia is linked to NP development21. In fact, the inhibition of both receptors alleviates NP symptoms22. A recent study implicates microglia and monocytes in the switch from acute to chronic pain23. Therefore, targeting microglia may be beneficial in future NP therapies. Similarly, research on depression mainly focuses on neuronal pathophysiology. Nevertheless, evidence links depression to the impairment of normal microglial structure and function24. Both high activation and a lack of microglial senescence are associated with depression-related impairment of neuroplasticity and neurogenesis2.
Kv1.3 Pivotal Role in Altered Microglial Function
Within the limited repertoire of potassium channels in leukocytes, the voltage-gated potassium channel Kv1.3 is crucial during the immune system response. Kv1.3 participates in physiological processes and in the onset of several pathophysiological dysfunctions in immune system cells25. Kv1.3, together with the calcium-dependent potassium channel KCa3.1, sustains the ionic driving force necessary for the T-cell activation. Moreover, proliferation and activation of macrophages is linked to an upregulation of Kv1.3. On the other hand, lupus erythematosus, psoriasis, type I diabetes and rheumatoid arthritis are associated with impaired Kv1.3 activity. In addition to autoimmune diseases, cancer, metabolic disorders and chronic inflammatory pathologies are linked to this channel26.
During the last twenty years, evidence about the Kv1.3 function in microglia reveal a strong association with neuropathology. Microglial activation leads to two different functional phenotypes: M1 (classic/proinflammatory) and M2 (alternative/neuroprotective)27. However, this functional polarization is under debate. While some researchers present clearly dichotomized results, others support a continuum of the activation status between the M1 and M2 phenotypes28. Some authors propose abandoning this oversimplification for a stimulus-based terminology. Similarly to the closely related macrophages29, Kv1.3 was functionally characterized in microglial cells and was strongly related to their proliferation30. The role of Kv1.3 in microglial activation was initially controversial and dependent on either the culture conditions or the mode of stimulation31. Some authors described that both classical M1 and an alternative M2 microglial subtype (M2a) exhibit an increase in Kv1.3 expression. Additionally, the M2a phenotype shows increases in anti-inflammatory genes and ROS production32. However, other lines of evidence indicate that Kv1.3 activity, together with KCa3.1, is confined exclusive to the M1 phenotype. On the other hand, cells in the M2 state have larger inward-rectifier potassium channel Kir2.1 currents than unstimulated microglial cells31,33. Concomitantly, Kv1.3 and KCa3.1 blockers impair the production of proinflammatory cytokines, such as IL-1? and TNF-α (Fig. 1). Moreover, activated microglia can kill neurons in a Kv1.3-dependent manner34.
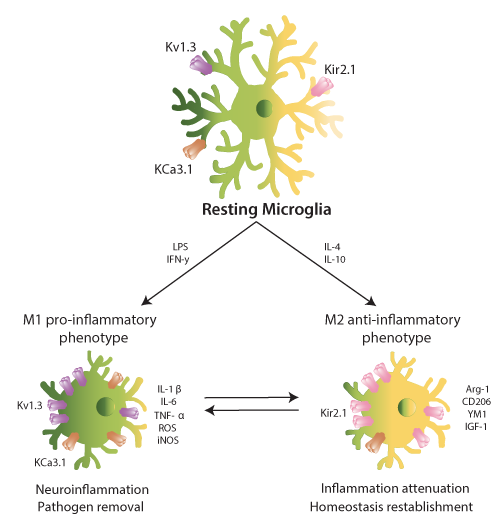
Figure 1: Microglial activation promotes a differential expression of ion channels. Resting microglia (top, center) express Kv1.3, KCa3.1 and Kir2.1. Upon stimulation with LPS or IFN-γ microglia polarizes to M1 pro-inflammatory phenotype (bottom, left), increasing the expression of Kv1.3 (LPS) and KCa3.1 (IFN-γ). M1-microglia releases cytokines and chemokines such as IL-1β and IL-6. These molecules mediate in neuroinflammation and pathogen removal. On the other hand, IL-4 and IL-10 enhance the M2 anti-inflammatory state (bottom, right), elevating the Kir2.1 expression. M2-microglia releases cytokines and chemokines such as Arg-1 and IGF-1. They mediate in inflammation attenuation and homeostasis reestablishment. The dichotomic phenotype is, nowadays, considered as a continuum for in vivo experiments.
Microglia is a central player in AD-associated neuroinflammation, and Kv1.3 regulates microglial function. Resting microglia also express Kv1.5, but upon activation, Kv1.3 expression increases at the plasma membrane30. Indeed, the exposure of rat microglial cells to Aβ increases Kv1.3 currents and leads to Kv1.3-dependent microglial priming35. Concomitantly, studies of the human brain pointed out that microglial cells from AD patients present increased expression of Kv1.336. Apart from microglial alterations, AD patients have Kv1.3 channels that are insensitive to glutamate-dependent modulation in T-cells. This condition results in a 4-fold increase in the Kv1.3 resting channel activity37. Indeed, AD treatment with N-methyl-D-aspartate receptor (NMDAR) antagonists, such as memantine, decreases the Kv1.3 T-cell conductivity38. In healthy donors, memantine also inhibits CD3 antibody-induced effects, impairs alloantigen-induced proliferation and suppresses the chemokine-induced migration of peripheral blood T-cells. CD4+ T-cells from AD patients receiving therapeutic doses of memantine exhibited a long-lasting reduced proliferative response to alloantigens39. Thus, inhibition of Kv1.3 in AD might exert a beneficial bipartite effect on T-cells and microglia. Actually, PAP-1, a Kv1.3 inhibitor, reduces neuroinflammation, decreases amyloid load, enhances hippocampal neuronal plasticity and improves behavioral deficits in a murine AD model. Concomitantly, PAP-1 stimulates an uptake by microglia40.
MS exhibits a tight link to T-cell malfunction. Evidence indicates that Kv1.3 blockers ameliorate MS symptoms via T-cell modulation26. Moreover, Kv1.3 expression increases in activated microglia, macrophages and reactive astrocytes at the peak of experimental autoimmune encephalomyelitis (EAE), a murine model of MS41. Indeed, the anti-Kv1.3 vaccination of EAE animals reduces pathogenic T-cells and increases protective T subsets, as well as decreases the infiltration of microglia/macrophages. After vaccine administration, microglial cells shift towards the M2 anti-inflammatory phenotype in the central nervous system42. The involvement of Kv1.3 has been further evaluated in other neuroinflammatory diseases. After status epilepticus, Kv1.3 expression increases in activated microglia43. Moreover, human microglia exhibits increased Kv1.3, KCa3.1 and Kir2.1 currents in brain stroke patients44. Therapeutically, the Kv1.3-inhibition diminishes the brain damage following a traumatic brain injury45. Therefore, a direct connection between microglia-mediated neuroinflammation and Kv1.3 expression is noticeable.
Due to its wide expression and prominent role during immune response, Kv1.3 blockers have been developed and tested as a potential treatment for diverse diseases. Studies in mice suggest Kv1.3 blockers as a possible therapy for obesity and insulin resistance. Moreover, Kv1.3 inhibition with Margatoxin reduces proliferative capacity of cancer cells. In EAE, the Kv1.3 channel blockage by ShK-22 ameliorated the clinical course of both moderate and severe EAE. Finally, two different treatments are being developed for treating psoriasis patients. Subcutaneous administration of dalatazatide (ShK-186) is demonstrated to improve the psoriatic skin lesions. Currently, a clinical trial is ongoing for studying the cutaneous effect of the Kv1.3 blocker PAP-126.
In this pathophysiological scenario, the modulation of Kv1.3 is crucial. Elevated Kv1.3 activity in the M1 microglial phenotype is linked to neuroinflammatory processes30,33. Kv1.3 is effectively targeted to the plasma membrane, thus controlling the membrane potential, which is crucial for cell activation. Therefore, the determinants responsible for Kv1.3 cell surface targeting are worth studying46. In this context, we have identified KCNE4, an endogenous β subunit in leukocytes that reduces Kv1.3 membrane abundance. KCNE4, initially named MinK-related peptide 3, is an auxiliary subunit that promiscuously regulates a wide variety of voltage-gated channels mostly functioning as a channel inhibitor47. KCNE4 mutations have been linked to several human immune system diseases, such as allergic rhinitis and acute lymphoblastic leukemia48,49. KCNE4, which is tightly regulated50, is highly expressed in mononuclear phagocytes, such as dendritic cells and macrophages, modulating Kv1.3. Thus, KCNE4, which fine-tunes Kv1.3 function, is an important regulator of leukocyte physiology. KCNE4 impairs Kv1.3 trafficking to the cell surface via two synergistic mechanisms (Fig. 2)47. Therefore, the pharmacological targeting of KCNE4 during neuroinflammation could decrease the harmful effects of the Kv1.3-dependent activation of leukocytes. Furthermore, limiting the effect to KCNE4-expressing cells might decrease treatment side-effects.
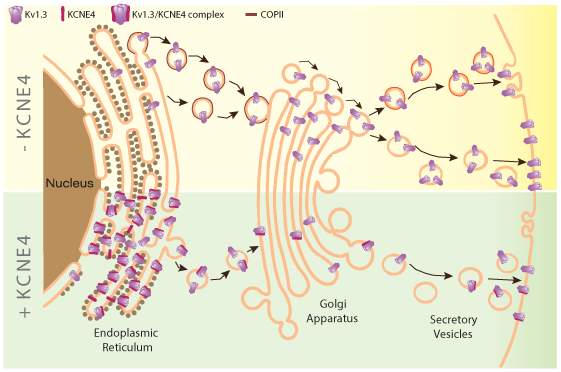
Figure 2: KCNE4 regulates Kv1.3 trafficking to the plasma membrane. Kv1.3 trafficking is highly efficient to the plasma membrane. The channel presents C-terminal export motifs recognized by Sec24, component of the COPII anterograde traffic machinery, at the endoplasmic reticulum (ER) which facilitates this process (- KCNE4, yellow top panel). Later in the secretory pathway, Kv1.3 will be directed to specific cell surface microdomains, such as lipid rafts and caveolae. KCNE4 interacting with Kv1.3 retains the channel at the ER impairing the complex localization at the plasma membrane (+ KCNE4, green bottom panel).
Concluding Remarks
Evidence demonstrates that the voltage-dependent Kv1.3 plays important roles in the cell physiology. Because this channel participates during the immune system response, a precise regulation of Kv1.3 activity and expression would fine-tune the magnitude of either the pro- or anti-inflammatory events. Microglia, being sentinel cells in the brain, functions as a first barrier against neuronal inflammation. Therefore, a deeper investigation of the Kv1.3 modulation by either a pharmacological targeting or by controlling the regulatory subunit expression in microglial cells is highly warranted to generate new treatment strategies50. Thereby, the control of Kv1.3 expression is a promising objective for improved treatment of neuroinflammatory diseases.
Acknowledgements
The work carried out by the Molecular Physiology Laboratory was funded by Ministerio de Economía y Competitividad (Spain) grants from the BFU (BFU2014-54928-R and BFU2017-87104-R) and Fondo Europeo de Desarrollo Regional (FEDER). The editorial assistance of the American Journal Experts is also acknowledged. The Molecular Physiology Laboratory would like to acknowledge all past members who have contributed to this research.
Footnote
SRR current address: Department of Physiology, Radboud University Medical Center, PO Box 9101, 6500 HB, Nijmegen, The Netherlands.
References
- Becher B, Spath S, Goverman J. Cytokine networks in neuroinflammation. Nat Rev Immunol. 2017; 17: 49-59.
- Skaper SD, Facci L, Zusso M, et al. An Inflammation-Centric View of Neurological Disease: Beyond the Neuron. Front Cell Neurosci. 2018; 12: 72.
- Paolicelli RC, Bolasco G, Pagani F, et al. Synaptic pruning by microglia is necessary for normal brain development. Science. 2011; 333: 1456-1458.
- Cuadrado A, Manda G, Hassan A, et al. Pharmacol Rev. 2018; 70(2): 348-383.
- Appel SH, Beers DR, Henkel JS. T cell-microglial dialogue in Parkinson's disease and amyotrophic lateral sclerosis: are we listening. Trends Immunol. 2010; 31: 7-17.
- Manocha GD, Floden AM, Rausch K, et al. APP Regulates Microglial Phenotype in a Mouse Model of Alzheimer's Disease. J Neurosci. 2016; 36: 8471-8486.
- Rivest S. Regulation of innate immune responses in the brain. Nat Rev Immunol. 2009; 9: 429-439.
- Heuberger RA. The frailty syndrome: a comprehensive review. J Nutr Gerontol Geriat. 2011; 30: 315-368.
- Biscaro B, Lindvall O, Tesco G, et al. Inhibition of microglial activation protects hippocampal neurogenesis and improves cognitive deficits in a transgenic mouse model for Alzheimer's disease. Neurodegener Dis. 2012; 9: 187-198.
- Zhang QS, Heng Y, Yuan YH, et al. Pathological alpha-synuclein exacerbates the progression of Parkinson's disease through microglial activation. Toxicol Lett. 2017; 265: 30-37.
- Schapansky J, Nardozzi JD, LaVoie MJ. The complex relationships between microglia, alpha-synuclein, and LRRK2 in Parkinson's disease. Neuroscience. 2015; 302:74-88.
- Turner MR, Cagnin A, Turkheimer FE, et al. Evidence of widespread cerebral microglial activation in amyotrophic lateral sclerosis: an [11C](R)-PK11195 positron emission tomography study. Neurobiol Dis. 2004; 15: 601-609.
- Casula M, Iyer AM, Spliet SG, et al. Toll-like receptor signaling in amyotrophic lateral sclerosis spinal cord tissue. Neuroscience. 2011; 179: 233-243.
- Jordan J, Segura T, Brea D, et al. Inflammation as therapeutic objective in stroke. Curr Pharm Design. 2008; 14: 3549-3564.
- Karve IP, Taylor JM, Crack PJ. The contribution of astrocytes and microglia to traumatic brain injury. Brit J of Pharmacol. 2016; 173: 692-702.
- Taib T, Leconte C, Van Steenwinckel J, et al. Neuroinflammation, myelin and behavior: Temporal patterns following mild traumatic brain injury in mice. PloS one. 2017; 12(9): e0184811.
- Johnson VE, Stewart JE, Begbie FD, et al. Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain. 2013; 136: 28-42.
- Loane DJ, Kumar A, Stoica BA, et al. Progressive neurodegeneration after experimental brain trauma: association with chronic microglial activation. Journal Neuropath Exp Neur. 2014; 73: 14-29.
- Ji RR, Xu ZZ, Gao YJ. Emerging targets in neuroinflammation-driven chronic pain. Nat Rev Drug Discov. 2014; 13: 533-548.
- Ren K, Dubner R. Interactions between the immune and nervous systems in pain. Nat Med. 2010; 16: 1267-1276.
- Burnstock G. Purinergic Mechanisms and Pain. Adv Pharmacol. 2016; 75: 91-137.
- Tsuda M. P2 receptors, microglial cytokines and chemokines, and neuropathic pain. J Neurosci Res. 2017; 95: 1319-1329.
- Peng J, Gu N, Zhou L, et al. Microglia and monocytes synergistically promote the transition from acute to chronic pain after nerve injury. Nat Commun. 2016; 7: 12029.
- Prinz M, Priller J. Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nat Rev Neurosci. 2014; 15: 300-312.
- Serrano-Albarras A., Estadella I, Cirera-Rocosa S, et al. Kv1.3: a multifunctional channel with many pathological implications. Expert Opin Ther Tar. 2018; 22: 101-105.
- Perez-Verdaguer M, Capera J, Serrano-Novillo C, et al. The voltage-gated potassium channel Kv1.3 is a promising multitherapeutic target against human pathologies. Expert Opin Ther Tar. 2016; 20: 577-591.
- Tang Y, Le W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol Neurobiol. 2016; 53: 1181-1194.
- Peferoen L, Kipp M, van der Valk P, et al. Oligodendrocyte-microglia cross-talk in the central nervous system. Immunology. 2014; 141: 302-313.
- Villalonga N, Ferreres JC, Argiles JM, et al. Potassium channels are a new target field in anticancer drug design. Recent pat Anti-Canc. 2007; 2: 212-223.
- Kettenmann H, Hanisch UK, Noda M, et al. Physiology of microglia. Physiol Rev. 2011; 91: 461-553.
- Nguyen HM, Blomster LV, Christophersen P, et al. Potassium channel expression and function in microglia: Plasticity and possible species variations. Channels (Austin). 2017; 11: 305-315.
- Siddiqui TA, Lively S, Schlichter LC. Complex molecular and functional outcomes of single versus sequential cytokine stimulation of rat microglia. J Neuroinflamm. 2016; 13: 66.
- Nguyen HM, Grössinger EM, Horiuchi M, et al. Differential Kv1.3, KCa3.1, and Kir2.1 expression in "classically" and "alternatively" activated microglia. Glia. 2017; 65: 106-121.
- Fordyce CB, Jagasia R, Zhu X, et al. Microglia Kv1.3 channels contribute to their ability to kill neurons. J Neurosci. 2005; 25: 7139-7149.
- Schilling T, Eder C. Amyloid-beta-induced reactive oxygen species production and priming are differentially regulated b ion channels in microglia. J Cell Physiol. 2011; 226: 3295-3302.
- Rangaraju S, Gearing M, Jin LW, et al. Potassium channel Kv1.3 is highly expressed by microglia in human Alzheimer's disease. J Alzheimers Dis. 2015; 44: 797-808.
- Poulopoulou C, Markakis I, Davaki P, et al. Aberrant modulation of a delayed rectifier potassium channel by glutamate in Alzheimer's disease. Neurobio Dis. 2010; 37: 339-348.
- Kahlfuβ S, Simma N, Mankiewics J, et al. Immunosuppression by N-methyl-D-aspartate receptor antagonists is mediated through inhibition of Kv1.3 and KCa3.1 channels in T cells. Mol Cell Biol. 2014; 34: 820-831.
- Lowinus T, Bose T, Busse S, et al. Immunomodulation by memantine in therapy of Alzheimer's disease is mediated through inhibition of Kv1.3 channels and T cell responsiveness. Oncotarget. 2016; 7: 53797-53807.
- Maezawa I, Nguyen HM, Di lucente J, et al. Kv1.3 inhibition as a potential microglia-targeted therapy for Alzheimer's disease: preclinical proof of concept. Brain. 2018; 141: 596-612.
- Bozic I, Tesovic K, Laketa D, et al. Voltage gated potassium channel Kv1.3 is upregulated on activated astrocytes in experimental autoimmune encephalomyelitis. Neurochem Res. 2018; 43: 1020-1034.
- Fan C, Longm R, You Y, et al. A novel PADRE-Kv1.3 vaccine effectively induces therapeutic antibodies and ameliorates experimental autoimmune encephalomyelitis in rats. Clin Immunol. 2018; pii: S1521-6616(17)30913-0.
- Menteyne A, Levavasseur F, Audinat E, et al. Predominant functional expression of Kv1.3 by activated microgliaof the hippocampus after Status epilepticus. PloS one. 2009; 4: e6770.
- Chen YJ, Nguyen HM, Maezawa I, et al. The potassium channel KCa3.1 constitutes a pharmacological target for neuroinflammation associated with ischemia/reperfusion stroke. J Cereb Blood Flow Metab. 2016; 36: 2146-2161.
- Reeves TM, Trimmer PA, Colley BS, et al. Targeting Kv1.3 channels to reduce white matter pathology after traumatic brain injury. Exp Neurol. 2016; 283: 188-203.
- Martinez-Marmol R, Pérez-Verdaguer M, Roig SR, et al. A non-canonical di-acidic signal at the C-terminus of Kv1.3 determines anterograde trafficking and surface expression. J Cell Sci. 2013; 126: 5681-5691.
- Sole L, Roig SR, Vallejo-Gracia A, et al. The C-terminal domain of Kv1.3 regulates functional interactions with the KCNE4 subunit. J Cell Sci. 2016; 129: 4265-4277.
- Freldin MB, Bragina EIu, Fedorova OS, et al. [Genome-wide association study of allergic diseases in Russians of Western Siberia]. Mol Biol (Mosk). 2011; 45(3):464-72.
- Treviño LR, Yang W, French D, et al. Germline genomic variants associated with chldhood acute lymphoblastic leukemia. Nat Genet. 2009; 41(9):1001-5.
- Sole L, Vallego-Gracia A, Roig SR, et al. KCNE gene expression is dependent on the proliferation and mode of activation of leukocytes. Channels (Austin). 2013; 7: 85-96.