
Neonatal Hypoxic-Ischemic Brain Injury: Apoptotic and Non-Apoptotic Cell Death
Qing Lu, Stephen M. Black
Abstract
The neuronal cell death associated with perinatal asphyxia, or hypoxic-ischemic (HI) brain injury, plays an important role in neonatal mortality and neurodevelopment retardation. The types of cell death associated with HI in the brain have been classified as being either apoptotic or necrotic. Here we describe the recent discoveries of multiple non-apoptotic cell death pathways: necroptosis; ferroptosis; and autosis (autophagy). These new cell death pathways expand our understanding of the mechanisms underlying the cell death associated with perinatal asphyxia. By targeting specific regulators of these pathways, new therapies may be developed that could protect the neonatal brain from the HI mediated injury.
Introduction
The neuronal cell death associated with neonatal hypoxic-ischemic (HI) brain injury is one of the major causes of neonatal mortality and neurodevelopment retardation. The mortality of HI brain injury is 30-40%. Significant neurological disorders and lifelong disabilities, such as cerebral palsy, seizures, visual impairment, mental retardation, learning impairment and epilepsy, occur in 20-40% of survivors1. Excitotoxicity, oxidative stress, as well as inflammation play important roles in the mechanism of neonatal HI brain injury. The cell death in HI injury, principally involves the cerebral cortex, hippocampus, cerebellar cortex, basal ganglia/thalamus, brainstem nuclei, and anterior horn cells of the spinal cord. The neuronal injury is influenced by the gestational age, cerebral blood supply, and the duration of the insult2. Investigations have shown that the types of cell death of neonatal brain injury are variable. Three types of cell death process have been commonly classified: apoptosis, necrosis and autophagy3,4. More recently three new non-apoptotic cell deaths pathways have been described: necroptosis, ferroptosis, and autosis (autophagy). In this review we will briefly describe how different types of cellular death impact ischemic brain injury in the neonate.
Apoptosis
The apoptotic pathway has been extensively documented in multiple animal models of neonatal brain injury5-7. Increased caspase-3 activation has been identified in brain sections in children who died after experiencing HI8. As apoptosis is also involved in normal brain development9, the neonatal brain may be more susceptible to this cell death pathway than the adult10,11. Apoptosis is thought to account for a significant portion of the neuronal cell loss associated with neonatal HI events. For example in the CA1 region of hippocampus, apoptotic cells account for 18-, 35- and 43-% of total cell numbers at 4-, 8- and 24-h after OGD (oxygen glucose deprivation)12. The apoptotic cell death pathway is histologically characterized by cytoplasmic condensation, nuclear pyknosis, chromatin condensation and fragmentation, cell rounding, membrane blebbing, cytoskeletal collapse, and the formation of membrane bound apoptotic bodies that are rapidly phagocytosed and digested by macrophages or neighboring cells. Oxidative stress plays an important role in induction of apoptosis after HI13.The key molecular players in the apoptotic pathway are APAF1, BAX, BAK, cytochrome c, as well as caspase-3. Mitochondrial dysfunction also plays a central role (Fig.1 b). Caspase-3 is cleaved in neurons within the ischemic core and penumbra, both early after reperfusion, and in the following hours and days. This suggests that caspase-3 participates in the delayed ischemic cell death associated with neonatal HI14. Selective caspase-3 inhibition has been shown to reduce HI brain injury in neonatal rodent models15-17. However, caspase-3−/− mice exhibit increased vulnerability to neonatal HI injury18, indicating that a non-caspase-3 dependent neuronal cell death pathway is involved or that caspase 3 also plays a protective role that is as yet undefined.
Necrosis
Despite numerous publications focusing on the apoptotic pathway, it should be emphasized that the majority of the cell death is necrosis (or unclassified types of cell death)19. Necrosis has been described as involving cytoplasmic swelling, nuclear dissolution and lysis. Necrosis is the major cell death phenotype in brain following HI injury in the neonatal rat. Within a few hours, the majority of neurons develop pyknotic nuclei and clear or eosinophilic perikarya. After 24h, these changes evolve into coagulation necrosis (infarction). A few days later, infarcts became partially cavitated, and by 3-weeks a smooth-walled cystic infarct develops20. Thus, necrotic cell death has a prolonged role in the progression of brain injury and represents a prevalent type of cell death after neonatal HI21. Despite its substantial involvement in the pathological process, little effort has gone into therapeutic targeting of necrotic cell death, due to the lack of identified markers and regulatory pathways. This large gap in our knowledge regarding necrotic cell death may also be an opportunity to develop new therapeutic targets to reduce neonatal HI brain injury. Encouragingly, substantial progress has been made in recent years to broaden our knowledge of the mechanisms underlying the neuronal cell death associated with neonatal HI that hopefully will lead to new approaches to protect the neonatal brain.
Alternative path triggering cell death, to eliminate damaged or dysfunction cells, independent of apoptosis have also been discovered. Mutant mice deficient in key apoptotic factors, such as caspase or Bcl-2, can survive to adult22,23. Non-apoptotic cell death has also been observed in neonatal HI brain injury model18. The regulation of several non-apoptotic cell death pathways such as necroptosis, autosis, and ferroptosis have been recently reported and interest in their role in brain injury is growing rapidly.
Necroptosis
That the stimulation of Fas /TNFα receptor (death receptor) family triggers a canonical apoptotic pathway, including the activation of multiple caspases is well established24. However, it has been shown that under apoptosis-deficient conditions Fas /TNFα can still induce non-apoptotic cell death (Fig.1 a). In this situation, TNF exposed cells were observed to undergo swelling and lysis25, with features of morphological injury that included low density vacuoles but without chromatin condensation26. More recently it has been shown that these vesicles are double membrane-enclosed, and filled with electron dense material27. The formation of these vesicles can be blocked using autophagy inhibitors. The term necroptosis is used to describe this alternative non-apoptotic cell death pathway. A specific necroptosis inhibitor, Nec-1 has been found through chemical library screening. Nec-1 efficiently inhibits TNFα-induced cell death, without the requiring caspase inhibition. Necroptosis has also been shown to occur in the injured mouse brain after middle cerebral artery occlusion (MCAO), with Nec-1 treatment significantly reducing infarct size27. Nec-1 has also been shown to improve motor function, Morris maze performance, and spatial memory in rats exposed to MCAO28. Nec-1 has also been shown to decrease injury in the forebrain and thalamus and protection correlated with decreased necrotic cell death and increased apoptotic cell death29. Nec-1 also reduces other injurious factors associated with HI injury including NO generation, protein nitration, and glutathione oxidation30. More specifically, studies have suggested that the receptor-associated adaptor RIP1 is the primary cellular target of Nec-131. Furthermore, the death receptor is not the only activator of necroptosis. A recent study found that deletion of the acid-sensing ion channel 1a (Asic1a) gene attenuates RIP1 phosphorylation and brain injury, suggesting ASIC1a-mediated RIP1 activation is also an important pathway in ischemic neuronal cell death32. This new form of neuronal necroptosis induced by extracellular acidosis is exciting as HI induces extracellular acidosis, via an increase in lactate release33, and opens up potential new avenues for both research into acid-induced neuronal cell death with the potential for new therapeutic targets being developed.
Ferroptosis
Besides necroptosis, another non-apoptotic cell death, that is iron-dependent, has recently been reported and named ferroptosis. Ferroptosis appears to be peroxidation-driven and requires abundant and accessible cellular iron (Fig.1 c). Ferroptosis was identified in cells under conditions where caspase, BAX and BAK were suppressed to attenuate apoptosis and RIP1/RIP3 expression decreased to reduce necroptosis34. Available data suggests that the lipid repair enzyme, GPX4 (glutathione peroxidase-4), is involved in this signal pathway. In the conditional ablation of GPX4 in neurons of mice, selective and rapid motor neuron degeneration is induced through ferroptosis, with no caspase-3 activation and TUNEL staining present35. Ferrostatin-1 has been identified as a small molecule inhibitor of ferroptosis, and significantly reduces the glutamate-induced cell death in mouse hippocampal slice cultures36. NADPH oxidase generated ROS likely promotes ferroptosis cell death36. Compared to apoptosis induced by staurosporine, necrosis caused by H2O2 or autophagy induced by rapamycin, ferroptosis is characterized by increased mitochondrial membrane density. The role of iron in ferroptosis remains unclear. A recent study has demonstrated that hydroxyl radical generated from iron accumulation, induces neonatal cell death, through a mechanism that requires NO and Iron regulatory protein-1 (IRP-1)37. As the lipid metabolism of the developing brain is poorly understood, more investigations are needed to unravel the complex role of lipid peroxidation in neonatal HI brain injury. Recent studies have also shown that HI attenuates β-oxidation via the disruption of carnitine homeostasis and subsequently mitochondrial function38. Studies in the neonatal dog also suggest that fatty acid oxidation may contribute up to 25% of the metabolites entering the TCA cycle39. Iron is intimately involved in fatty acid oxidation, not only as a component of cytochromes within the electron transport chain, but because it is involved in the production of carnitine40,41. Further, iron deficiency during pregnancy may also affect neonatal brain lipid metabolism and myelination42.
Autosis
Autophagy refers to any cellular degradative pathway that involves the delivery of cytoplasmic cargo to the lysosome. It occurs at basal levels in most tissues and contributes to the protein turnover of cytoplasmic components. Adaptive autophagy exerts cytoprotective and anti-inflammatory effects43. Autophagy has been shown to be neuroprotective in some forms of acute brain damage, including methamphetamine intoxication, spinal cord injury and subarachnoid haemorrhage. However, the autophagic machinery precipitates a peculiar form of cell death (known as autosis) that appears to contribute to the development of other types of acute brain damage including neonatal asphyxia44.
Impressive progress on the autophagy-mediated cell death has been made recently. Autophagy induced cell death neither activates caspase 3, nor is prevented by genetic deletion of RIPK1 and RIPK3, making it distinct from apoptosis and necroptosis. The term "autosis" has been coined to define the cell death mediated by autophagy genes and is characterized by focal perinuclear swelling. Autophagy (autosis) involves the degradation of cellular components via the lysosome machinery. The major intracellular characteristics of autophagy are the sequestration of damaged proteins and dysfunctional organelles into the double-membrane structure, to form the autophagosome which is decorated with the autophagic protein LC3. The autophagosome subsequently fuses with lysosomes for degradation and recycling45. Multiple proteins are required for initiation, vesicle elongation, autophagosome maturation, lysosome fusion, and degradation. Targeting the autophagic machinery could have significant impact in protecting the neonatal brain from HI-mediated cell death. Although the generation of the autophagosome is not fully understood, several key regulators have been revealed. These include mTOR, AMPK, and the PI3K complex. In the initiation phase class III PI3 kinase complex is thought perform a key role along with VPS34, VSP15, ATG14L and Beclin1(Fig.1 d). This complex generates PI3P, which determines the site at which the double membrane subsequently elongates. The Beclin1-VPS34 initiation complex is controlled through a preinitiation complex, which is activated by AMPK and inhibited by mTOR46. During glucose deprivation, MAPK can also act directly on the Beclin1-VPS34 complex, by phosphorylating Beclin1 at S91/S94, and T163 /S165 in VPS34 to induce autophagy47. Accumulating evidence has shown that autophagy is associated with neuronal cell death in both neurodegenerative diseases48 and in neonatal HI brain injury. A dramatic increase in autophagosome formation, accompanied with extensive neuronal cell death, has been described in the hippocampal pyramidal region of neonatal mice exposed to HI and mice deficient in Atg7 (Autophagy-related protein 7) in the brain exhibit nearly complete protection against neuronal cell from death49. Intense vacuolization and autophagosome formation have been observed the hippocampus CA1 and CA3 regions in a neonatal rat HI brain injury model50. Autophagy protein expression and autophagosome generation have also been reported in hippocampal slice cultures exposed to OGD51. Interestingly, in the rat neonatal HI brain injury model, most of the neuronal cell death in the CA3 region appears to be through autophagy, with high numbers of autophagosomes, empty vacuoles, and focal ballooning of the perinuclear space being observed52. Importantly, significant LC3 staining, as an autophagy marker, has been found in the brains of human neonates53. Thus, accumulating evidence implicates autophagy as an important player in the neuronal cell death associated with neonatal HI injury brain. Cardiac glycosides have been identified to be potent inhibitors of autosis. One such glycoside, neriifolin blocks the increase in autophagy induced by HI in the brain, and was found to be highly protective against HI-mediated brain injury52. In addition, new autophagy activators are being developed for therapeutic purposes. For example, the increasing autophagy using fluendazole, interrupts HIV transmission54. Thus, targeting the autophagy pathway has a potential therapeutic role in the spectrum of human disease. 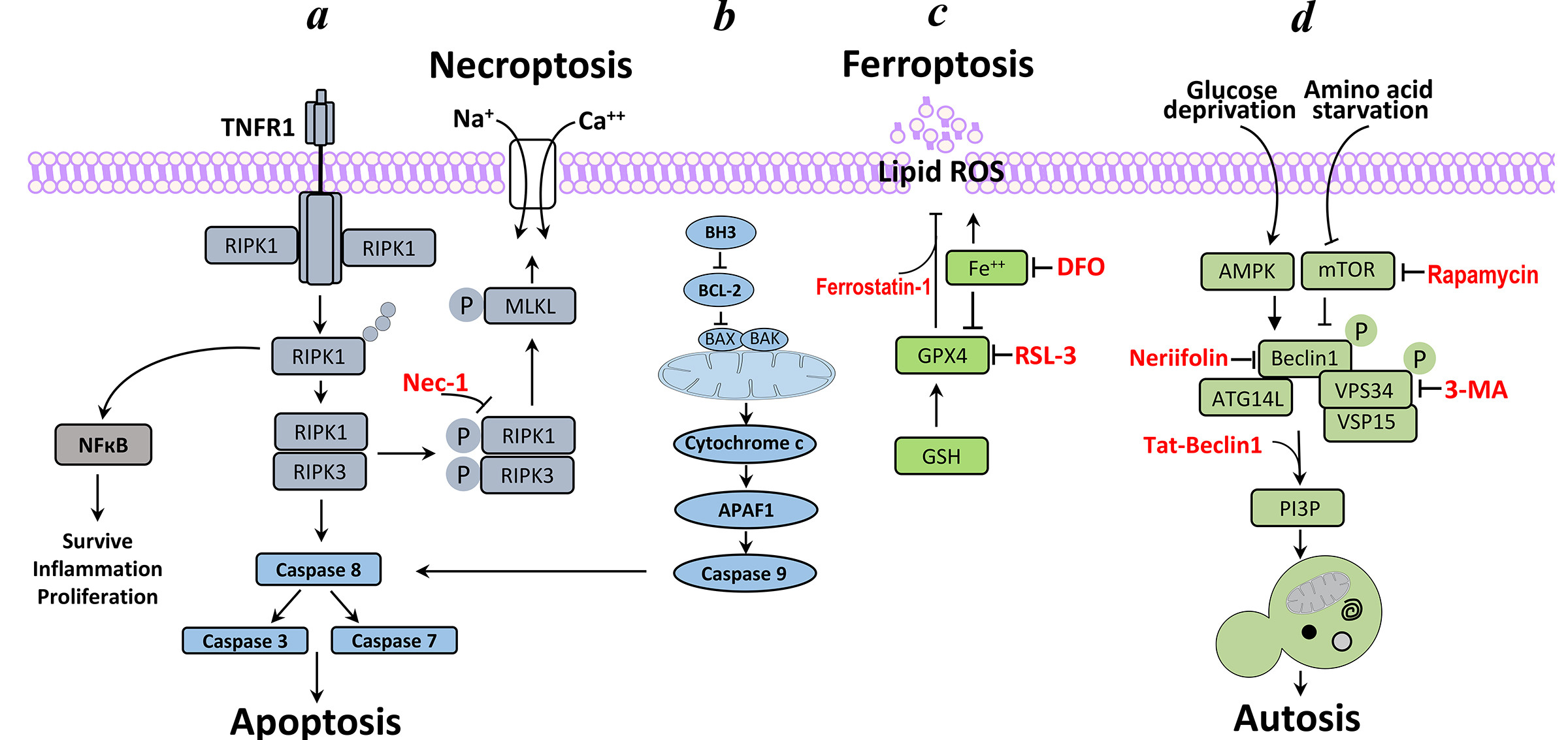
Figure 1:Neuronal cell death pathways in the neonatal hypoxic-ischemic brain injury.
a. Necroptosis-Ligation of tumour necrosis factor receptor 1 (TNFR1) results in the recruitment of receptor-interacting serine/threonine protein kinase 1 (RIPK1). When caspase 8 is deleted or inhibited, RIPK1 interacts with RIPK3, to form the necrosome; this interaction can be inhibited by necrostatin 1 (NEC1). RIPK3 recruits and phosphorylates (P) mixed-lineage kinase domain-like protein (MLKL), leading to the formation of MLKL oligomers that translocate to the plasma membrane. Once at the plasma membrane, MLKL forms membrane-disrupting pores, which regulate the influx of Na+ and Ca2+, resulting in membrane rupture. b. Apoptosis- Cell commitment to apoptosis is regulated by the interplay between the anti-apoptotic BCL-2 subfamily of proteins and the pro-apoptotic BH3-only subfamily. During apoptosis, BH3-only proteins facilitate a BAX (BCL-2-associated X protein)- and BAK (BCL-2 antagonist/killer)-dependent release of cytochrome c, which binds to APAF1 and gives rise to the apoptosome, which, in turn, stimulates the executioner caspases 3 and 7. c. Ferroptosis-GPX4 (glutathione peroxidase-4) is the key regulator of the ferroptosis pathway, inducing iron dependent lipid peroxidation. DFO (deforxamine) an iron-chelating agent; RSL3, RAS-selective lethal compound block feroptosis. d. Autosis (autophagy)-The upstream kinases that sense cellular nutrient and energy status exert inhibitory and stimulatory effects on the Class III PI3K complex. Bcl-2 and a variety of other proteins are also involved in this step (but for simpliciaty are not presented). The Class III PI3K complex generates PI3P leading to the vesicle elongation reaction.
Conclusion
The recent discovery of multiple non-apoptotic cell death pathways has expanded our understanding of the mechanism underlying the neuronal cell death associated with neonatal HI brain injury. Compounds are already being generated from small molecules screening or using designed peptides that can active or inhibit these processes. It is hoped that by targeting specific regulators of these new pathways, new therapies will be developed that are able to protect the neonatal brain.
References
- Juul SE, Ferriero DM (2014) Pharmacologic neuroprotective strategies in neonatal brain injury. Clinics in perinatology 41:119-131.
- Swaiman KF, Ashwal S (1999) Pediatric Neurology Principles & Practice. Mosby.
- Northington FJ, Ferriero DM, Flock DL, Martin LJ (2001) Delayed neurodegeneration in neonatal rat thalamus after hypoxia-ischemia is apoptosis. The Journal of neuroscience : the official journal of the Society for Neuroscience 21:1931-1938.
- Zille M, Farr TD, Przesdzing I, Muller J, Sommer C, Dirnagl U, Wunder A (2012) Visualizing cell death in experimental focal cerebral ischemia: promises, problems, and perspectives. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism 32:213-231.
- Nakajima W, Ishida A, Lange MS, Gabrielson KL, Wilson MA, Martin LJ, Blue ME, Johnston MV (2000) Apoptosis has a prolonged role in the neurodegeneration after hypoxic ischemia in the newborn rat. The Journal of neuroscience : the official journal of the Society for Neuroscience 20:7994-8004.
- Manabat C, Han BH, Wendland M, Derugin N, Fox CK, Choi J, Holtzman DM, Ferriero DM, Vexler ZS (2003) Reperfusion differentially induces caspase-3 activation in ischemic core and penumbra after stroke in immature brain. Stroke; a journal of cerebral circulation 34:207-213.
- Pulera MR, Adams LM, Liu H, Santos DG, Nishimura RN, Yang F, Cole GM, Wasterlain CG (1998) Apoptosis in a neonatal rat model of cerebral hypoxia-ischemia. Stroke; a journal of cerebral circulation 29:2622-2630.
- Rossiter JP, Anderson LL, Yang F, Cole GM (2002) Caspase-3 activation and caspase-like proteolytic activity in human perinatal hypoxic-ischemic brain injury. Acta neuropathologica 103:66-73.
- Raff MC, Barres BA, Burne JF, Coles HS, Ishizaki Y, Jacobson MD (1993) Programmed cell death and the control of cell survival: lessons from the nervous system. Science 262:695-700.
- Ferriero DM, Miller SP (2010) Imaging selective vulnerability in the developing nervous system. Journal of anatomy 217:429-435.
- Cheng Y, Deshmukh M, D'Costa A, Demaro JA, Gidday JM, Shah A, Sun Y, Jacquin MF, Johnson EM, Holtzman DM (1998) Caspase inhibitor affords neuroprotection with delayed administration in a rat model of neonatal hypoxic-ischemic brain injury. The Journal of clinical investigation 101:1992-1999.
- Nakatomi H, Kuriu T, Okabe S, Yamamoto S, Hatano O, Kawahara N, Tamura A, Kirino T, Nakafuku M (2002) Regeneration of hippocampal pyramidal neurons after ischemic brain injury by recruitment of endogenous neural progenitors. Cell 110:429-441.
- Chen H, Yoshioka H, Kim GS, Jung JE, Okami N, Sakata H, Maier CM, Narasimhan P, Goeders CE, Chan PH (2011) Oxidative stress in ischemic brain damage: mechanisms of cell death and potential molecular targets for neuroprotection. Antioxidants & redox signaling 14:1505-1517.
- Namura S, Zhu J, Fink K, Endres M, Srinivasan A, Tomaselli KJ, Yuan J, Moskowitz MA (1998) Activation and cleavage of caspase-3 in apoptosis induced by experimental cerebral ischemia. The Journal of neuroscience : the official journal of the Society for Neuroscience 18:3659-3668.
- Han BH, Xu D, Choi J, Han Y, Xanthoudakis S, Roy S, Tam J, Vaillancourt J, Colucci J, Siman R, Giroux A, Robertson GS, Zamboni R, Nicholson DW, Holtzman DM (2002) Selective, reversible caspase-3 inhibitor is neuroprotective and reveals distinct pathways of cell death after neonatal hypoxic-ischemic brain injury. The Journal of biological chemistry 277:30128-30136.
- Han BH, DeMattos RB, Dugan LL, Kim-Han JS, Brendza RP, Fryer JD, Kierson M, Cirrito J, Quick K, Harmony JA, Aronow BJ, Holtzman DM (2001) Clusterin contributes to caspase-3-independent brain injury following neonatal hypoxia-ischemia. Nat Med 7:338-343.
- Arvin KL, Han BH, Du Y, Lin SZ, Paul SM, Holtzman DM (2002) Minocycline markedly protects the neonatal brain against hypoxic-ischemic injury. Annals of neurology 52:54-61.
- West T, Atzeva M, Holtzman DM (2006) Caspase-3 deficiency during development increases vulnerability to hypoxic-ischemic injury through caspase-3-independent pathways. Neurobiology of disease 22:523-537.
- Northington FJ, Chavez-Valdez R, Martin LJ (2011) Neuronal cell death in neonatal hypoxia-ischemia. Annals of neurology 69:743-758.
- Towfighi J, Zec N, Yager J, Housman C, Vannucci RC (1995) Temporal evolution of neuropathologic changes in an immature rat model of cerebral hypoxia: a light microscopic study. Acta neuropathologica 90:375-386.
- Carloni S, Carnevali A, Cimino M, Balduini W (2007) Extended role of necrotic cell death after hypoxia-ischemia-induced neurodegeneration in the neonatal rat. Neurobiology of disease 27:354-361.
- Lindsten T, Thompson CB (2006) Cell death in the absence of Bax and Bak. Cell Death Differ 13:1272-1276.
- Los M, Wesselborg S, Schulze-Osthoff K (1999) The role of caspases in development, immunity, and apoptotic signal transduction: lessons from knockout mice. Immunity 10:629-639.
- Wallach D, Varfolomeev EE, Malinin NL, Goltsev YV, Kovalenko AV, Boldin MP (1999) Tumor necrosis factor receptor and Fas signaling mechanisms. Annual review of immunology 17:331-367.
- Vercammen D, Vandenabeele P, Beyaert R, Declercq W, Fiers W (1997) Tumour necrosis factor-induced necrosis versus anti-Fas-induced apoptosis in L929 cells. Cytokine 9:801-808.
- Kawahara A, Ohsawa Y, Matsumura H, Uchiyama Y, Nagata S (1998) Caspase-independent cell killing by Fas-associated protein with death domain. The Journal of cell biology 143:1353-1360.
- Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J (2005) Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nature chemical biology 1:112-119.
- You Z, Savitz SI, Yang J, Degterev A, Yuan J, Cuny GD, Moskowitz MA, Whalen MJ (2008) Necrostatin-1 reduces histopathology and improves functional outcome after controlled cortical impact in mice. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism 28:1564-1573.
- Northington FJ, Chavez-Valdez R, Graham EM, Razdan S, Gauda EB, Martin LJ (2011) Necrostatin decreases oxidative damage, inflammation, and injury after neonatal HI. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism 31:178-189.
- Chavez-Valdez R, Martin LJ, Flock DL, Northington FJ (2012) Necrostatin-1 attenuates mitochondrial dysfunction in neurons and astrocytes following neonatal hypoxia-ischemia. Neuroscience 219:192-203.
- Degterev A, Hitomi J, Germscheid M, Ch'en IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, Hedrick SM, Gerber SA, Lugovskoy A, Yuan J (2008) Identification of RIP1 kinase as a specific cellular target of necrostatins. Nature chemical biology 4:313-321.
- Wang YZ, Wang JJ, Huang Y, Liu F, Zeng WZ, Li Y, Xiong ZG, Zhu MX, Xu TL (2015) Tissue acidosis induces neuronal necroptosis via ASIC1a channel independent of its ionic conduction. Elife 4.
- Ljunggren B, Norberg K, Siesjo BK (1974) Influence of tissue acidosis upon restitution of brain energy metabolism following total ischemia. Brain research 77:173-186.
- Yang WS, Stockwell BR (2008) Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chemistry & biology 15:234-245.
- Chen L, Hambright WS, Na R, Ran Q (2015) Ablation of the Ferroptosis Inhibitor Glutathione Peroxidase 4 in Neurons Results in Rapid Motor Neuron Degeneration and Paralysis. The Journal of biological chemistry 290:28097-28106.
- Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, Morrison B, 3rd, Stockwell BR (2012) Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149:1060-1072.
- Lu Q, Harris VA, Rafikov R, Sun X, Kumar S, Black SM (2015) Nitric oxide induces hypoxia ischemic injury in the neonatal brain via the disruption of neuronal iron metabolism. Redox Biol 6:112-121.
- Rau TF, Lu Q, Sharma S, Sun X, Leary G, Beckman ML, Hou Y, Wainwright MS, Kavanaugh M, Poulsen DJ, Black SM (2012) Oxygen glucose deprivation in rat hippocampal slice cultures results in alterations in carnitine homeostasis and mitochondrial dysfunction. PloS one 7:e40881.
- Spitzer JJ (1975) Application of tracers in studying free fatty acid metabolism of various organs in vivo. Federation proceedings 34:2242-2245.
- Lindstedt G (1967) Hydroxylation of gamma-butyrobetaine to carnitine in rat liver. Biochemistry 6:1271-1282.
- Hulse JD, Ellis SR, Henderson LM (1978) Carnitine biosynthesis. beta-Hydroxylation of trimethyllysine by an alpha-ketoglutarate-dependent mitochondrial dioxygenase. The Journal of biological chemistry 253:1654-1659.
- Dobbing J (1990) Iron deficiency disease in infants. Springer,
- Kroemer G, Marino G, Levine B (2010) Autophagy and the integrated stress response. Molecular cell 40:280-293
- Galluzzi L, Bravo-San Pedro JM, Blomgren K, Kroemer G (2016) Autophagy in acute brain injury. Nat Rev Neurosci
- Choi AM, Ryter SW, Levine B (2013) Autophagy in human health and disease. The New England journal of medicine 368:651-662
- Green DR, Levine B (2014) To be or not to be? How selective autophagy and cell death govern cell fate. Cell 157:65-75
- Kim J, Kim YC, Fang C, Russell RC, Kim JH, Fan W, Liu R, Zhong Q, Guan KL (2013) Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell 152:290-303
- Rubinsztein DC (2006) The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 443:780-786
- Koike M, Shibata M, Tadakoshi M, Gotoh K, Komatsu M, Waguri S, Kawahara N, Kuida K, Nagata S, Kominami E, Tanaka K, Uchiyama Y (2008) Inhibition of autophagy prevents hippocampal pyramidal neuron death after hypoxic-ischemic injury. Am J Pathol 172:454-469
- Sheldon RA, Hall JJ, Noble LJ, Ferriero DM (2001) Delayed cell death in neonatal mouse hippocampus from hypoxia-ischemia is neither apoptotic nor necrotic. Neuroscience letters 304:165-168
- Lu Q, Harris VA, Kumar S, Mansour HM, Black SM (2015) Autophagy in neonatal hypoxia ischemic brain is associated with oxidative stress. Redox Biol 6:516-523
- Liu Y, Shoji-Kawata S, Sumpter RM, Jr., Wei Y, Ginet V, Zhang L, Posner B, Tran KA, Green DR, Xavier RJ, Shaw SY, Clarke PG, Puyal J, Levine B (2013) Autosis is a Na+,K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proceedings of the National Academy of Sciences of the United States of America 110:20364-20371
- Xie C, Ginet V, Sun Y, Koike M, Zhou K, Li T, Li H, Li Q, Wang X, Uchiyama Y, Truttmann AC, Kroemer G, Puyal J, Blomgren K, Zhu C (2016) Neuroprotection by selective neuronal deletion of ATG7 in neonatal brain injury. Autophagy 12:410-423
- Chauhan S, Ahmed Z, Bradfute SB, et al. Pharmaceutical screen identifies novel target processes for activation of autophagy with a broad translational potential. Nature communications. 2015; 6:8620