
New Insights on Neuronal Functions of Ghrelin Receptor GHS-R in Obesity
Jong Han Lee1,2, Sahar Eshghjoo3, Jon Davis4, Robert C. Alaniz3, Yuxiang Sun2,5*
1College of Pharmacy, Gachon University, Incheon, Gyeonggi, Korea
2Children’s Nutrition Research Center, Department of Pediatrics, Baylor College of Medicine, Houston, TX, USA
3Department of Microbial Pathogenesis and Immunology, Texas A&M University, Health Science Center, College Station, TX, USA
4Integrative Physiology and Neuroscience, Washington State University, Pullman WA, USA
5Department of Nutrition and Food Science, Texas A&M University, College Station, TX, USA
Abstract
Obesity is defined as excessive fat accumulation caused by the imbalance of energy intake versus expenditure. Low-grade chronic inflammation is a hallmark of obesity, and it is closely linked to insulin resistance. Obesity-induced inflammation and its associated metabolic dysfunctions underlay pathological processes of many chronic diseases such as insulin resistance, diabetes, sarcopenia and cardiovascular disease. Ghrelin is the only known circulating orexigenic hormone; it stimulates growth hormone release, and increases adiposity and insulin resistance. Ghrelin’s functions are mediated through its receptor, growth hormone secretagogue receptor (GHS-R). The brain plays a critical role in energy homeostasis and energy metabolism. GHS-R is primarily expressed in the brain; the brain is a key ghrelin targeting site. The current review discusses the insights we have gained from global GHS-R knockout mice and neural conditional GHS-R knockout mice, specifically involving the roles of GHS-R in food intake, feeding adaptation, thermogenesis, meta-inflammation, and physical activity.
Obesity is the consequence of the imbalance of energy intake versus expenditure. It is characterized by increasing fat accumulation in adipose tissues and non-adipose tissues such as liver and muscle. Obesity-induced metabolic impairment promotes low grade chronic inflammation in a wide range of peripheral tissues; this metabolically-triggered inflammation is termed "meta-inflammation”1,2. Meta-inflammation elicits further metabolic dysfunctions in insulin targeting tissues such as adipose tissues, liver and muscle. The vicious cycle between metabolic dysfunction and inflammation underlies pathological processes of many chronic diseases such as obesity, insulin resistance, diabetes and cardiovascular disease. Thus, obesity is a major risk factor for the development of insulin resistance, type 2 diabetes, cardiovascular disease, and muscle dysfunction3, 4. Hypothalamus is a key site for the regulation of energy homeostasis; it senses the energy status of the body by receiving hormonal signals from ghrelin, leptin and insulin, and modulates feeding behavior and energy expenditure5, 6. Neuronal dysregulation in the hypothalamus is a major pathological hallmark of obesity7. Reducing energy intake (such as reducing food take) and/or increasing energy expenditure (such as increasing physical activity and/or non-shivering thermogenesis) offer promising strategies for combating obesity.
Ghrelin is the only known circulating orexigenic hormone associated with growth hormone release, meal initiation, adiposity and insulin resistance8. Ghrelin is ubiquitously expressed, and the highest expression is detected in the stomach and gut8. Ghrelin increases body weight by increasing food intake through its receptor, growth hormone secretagogue receptor (GHS-R)8. GHS-R is primarily expressed in the brain, with the highest expression in the hypothalamus and relatively lower expression in other brain regions8, 9. Low levels of GHS-R expression are also detectable in peripheral tissues such as macrophages, muscle, and white and brown adipose tissues10, 11. Studies have shown that circulating ghrelin and GHS-R expression in tissues are altered under the conditions of high-fat diet-feeding, obesity and aging9, 12. These findings suggest that ghrelin-GHS-R signaling may be involved in the regulation of energy homeostasis. The brain has a major role in energy regulation, and it is a key targeting site of ghrelin signaling. While we and others have published a number of papers using GHS-R global knockout mice13, 14, the neuron-specific roles of GHS-R and the specific site of actions of GHS-R in the brain remain unaddressed. To tackle this important question, we have generated two new GHS-R conditional knockout mice to investigate the functions of GHS-R in neurons. In this review, we will discuss the insights we have gained from both GHS-R global and neural conditional GHS-R knockout mice.
The Effect of GHS-R on Food Intake
GHS-R is expressed in orexigenic agouti-related peptide (AgRP)/neuropeptide Y (NPY), but not in anorexic pro-opiomelanocortin (POMC) neurons in the hypothalamus6. Ghrelin promotes food intake and increases body weight by simultaneously stimulating AgRP/NPY neurons and suppressing POMC neurons15, 16. Even though pharmacological administration of ghrelin has robust orexigenic effect, we found that total food intake of Ghsr-null mice does not change regardless of age or diet13,17,18. Interestingly, however, we found that Ghsr-null mice ate larger meals and had decreased feeding frequency19. We recently reported that ablation of GHS-R abolishes the ghrelin-induced excitatory effect on AgRP/NPY neurons, indicating that GHS-R is essential for ghrelin-induced excitation of AgRP/NPY neurons20. The inhibitory effect of ghrelin on POMC neurons is thought to be an indirect effect, mediated by increasing GABAergic synaptic input from NPY/AgRP neurons15, 16, 21. Interestingly, our study showed that blocking GABAergic receptors with gabazine increased the basal firing activity of POMC neurons in both wild-type and GHS-R null mice, and ghrelin excites POMC neurons of GHS-R null mice in the presence of gabazine20. Our new findings suggest that ghrelin may regulate POMC neurons through an unidentified mechanism, independent from GABAergic synaptic input into POMC neurons. Collectively, these findings support that GHS-R is essential for ghrelin-induced excitation of AgRP/NPY neurons, but not for ghrelin-induced suppression of POMC; GHS-R is not required for long-term total energy intake of the mice.
The Role of GHS-R on Feeding Adaptation
GHS-R is also expressed in dopaminergic (DAergic) neurons in the ventral tegmental area (VTA), albeit to a much lesser extent than that observed in the arcuate nucleus22. Notably, ghrelin administration directly into the VTA augments palatable food intake and food-seeking behaviors23, indicating that this population of receptors is central in control of critical aspects of feeding. It is hypothesized that ghrelin controls these functions through modulation of DAergic neuronal activity. Indeed, ghrelin application depolarizes VTA DAergic neurons24, an effect dependent on excitatory synaptic input to these cells that lends credibility to this notion. In a recent series of studies, we discovered that blockade of GHS-R signaling with the antagonist D-Lys-GHRP-6 abolished tonic DAergic firing in the VTA of male rats25. This finding indicates that GHS-R activity, through an unknown mechanism, controls dopamine firing independent of ghrelin. We also find that D-Lys-GHRP-6 treatment was ineffective to alter tonic DAergic firing in rats that received weight loss surgery, and these rats displayed low circulating ghrelin concentrations in the blood and reduced hedonic intake of palatable food25. Collectively, these studies suggest that the ghrelin ligand regulates GHS-R activity through alternative mechanisms to control palatable food intake and the feeding motivation.
GHS-R is a constitutively active G protein-coupled receptor (GPCR), which is able to initiate signaling activation in the absence of ligand ghrelin26, 27. GHS-R has the ability to form heterodimers with other GPCRs, such as dopamine receptors, melanocortin 3 receptors, and serotonin 2C receptors26, 28, 29, and these dimerization’s alter the neuronal circuits of feeding behavior26. It has been reported that the formation of heterodimers between GHS-R and dopamine receptor 2 modifies dopamine signaling, resulting in Gβγ subunit-dependent but calcium mobilization-independent GHS-R basal activation28. It was also fund that dopamine receptor 2 agonist suppresses appetite in both wild-type and ghrelin-null mice, but not in GHS-R null mice28. The data suggest that the dopamine-induced appetite suppression is GHS-R dependent, but not ghrelin-dependent. The functional significance and molecular mechanism of GHS-R constitutive activity needs to be further investigated.
The Role of GHS-R in Thermogenesis
Emerging evidence suggests that non-shivering thermogenesis in brown adipose tissue plays a crucial role in energy homeostasis in both rodents and humans30, 31. Thermogenic capacity in the body positively correlates with energy expenditure, but negatively correlates with body fat mass. Obesity reduces thermogenesis in brown adipose tissue (BAT) through down-regulation of mitochondrial biogenesis and dysregulation of mitochondrial dynamics, as shown by us and others12, 31, 32. Under a cold environment, the sympathetic nervous system (SNS) releases norepinephrine (NE), which in turn activates β 3-adrenergic receptor (β 3-AR) in brown adipocytes. The β 3-AR signaling activates intracellular signaling cascade, such as the protein kinase A (PKA)-hormone-sensitive lipase (HSL) axis, to increase thermogenesis by activation of mitochondrial uncoupling protein 1 (UCP1) in brown adipocytes. We have reported that aging GHS-R null mice are lean and insulin-sensitive, showing increased thermogenic activation in BAT12. In addition to BAT, studies also indicate that “beige” adipocytes in subcutaneous adipose tissues are also involved in thermogenesis31, 33. While BAT and subcutaneous adipose tissues are the sites for thermogenesis, the sympathetic nervous system (SNS) involving many brain regions is the driver of thermogenic activation34. To distinguish the roles of GHS-R in neurons from peripheral tissues, we generated a conditional knockout mouse line using neuron-specific driver of Synapsin-cre. Remarkably, we found that neuronal deletion of GHS-R completely prevents diet-induced obesity9. High-fat diet (HFD)-fed neuronal-specific GHS-R deficient young mice have increased expression of thermogenic regulators (UCP-1, UCP-3, PGC-1α and β3-AR) in both BAT and subcutaneous fat tissues under cold exposure9. Furthermore, our data indicate that neuronal GHS-R deletion regulates genes mediating central thermogenic activation in ventromedial hypothalamus (VMH), paraventricular nucleus (PVN) and lateral hypothalamus (LH), implying elevated sympathetic outflow to brown adipose tissue and “beige” subcutaneous adipose tissues9. To further identify the specific site that governs GHS-R mediated SNS-thermogenic regulation, we further generated a conditional knockout mouse line specifically targeting AgRP neurons35. Our data demonstrate that suppression of GHS-R in AgRP neurons mitigates diet-induced obesity by regulating thermogenic activity in brown and “beige” adipocytes35. However, the thermogenic phenotype and protective effect on diet-induced obesity of AgRP-specific GHS-R knockout mice was less pronounced when compared to that of GHS-R total neuronal knockout mice9, 35. This result indicates that neurons other than AgRP neurons likely contribute to centrally-mediated thermogenesis, and further studies are needed to identify the additional sites governing GHS-R mediated sympathetic activation.
The Role of GHS-R on Meta-Inflammation in Adipose Tissues
Metabolically-triggered "meta-inflammation" is a hallmark pathology of obesity and insulin resistance, and macrophages are a primary source of inflammatory effectors that contribute to meta-inflammation1. Macrophages are present as monocytes in the circulation, or as infiltrated or resident macrophages in the tissues36-39. Macrophages utilize both endocrine and paracrine mechanisms to elicit systemic and tissue-specific effects36-39. In response to environmental signals of various stimuli, macrophages undergo dynamic changes and reprogram to either pro-inflammatory or anti-inflammatory states2,39.
We showed that GHS-R expression in mature peritoneal macrophages is increased during aging39. We reported that GHS-R deletion reduces pro-inflammatory cytokine expression in white and brown adipose tissues, which suggests the GHS-R activation promotes pro-inflammatory polarization in macrophages39. Some studies suggest that ghrelin’s effect in macrophages may be mediated by scavenger receptor CD36 and peroxisome proliferator activated receptor γ (PPARγ)40, 41. Lucchi et.al showed that ghrelin activates PPARγ in hippocampus; intriguingly, ghrelin-induced PPARγ immunoreactivity in hippocampus remains intact in mice treated with GHS-R antagonist, and PPARγ antagonist counteracts the effect of GHS-R antagonist42. These findings suggest ghrelin and GHS-R may activate different signaling pathways in macrophages, and ghrelin’s effect in macrophages could be independent of GHS-R. We have also observed that GHS-R deletion attenuates dietary insult-induced inflammation and lipid accumulation in adipose tissue and liver18. Global Ghsr-null mice are protected against high fructose corn syrup (HFCS)-induced adipose inflammation and insulin resistance; the adipose tissue macrophages of Ghsr-null mice showed decreased M1, but increased M218. These results support that GHS-R contributes to meta-inflammation in adipose tissues by regulating macrophage polarization and macrophage-associated adipose tissue inflammation and insulin resistance.
The Role of GHS-R on Physical Activity
Physical inactivity and sedentary lifestyle are crucial determinants for being overweight or obese in a Western population, and the incidences of obesity are significantly higher in aging population43. Exercise is thought to be an effective treatment of obesity in humans and rodents by increasing energy expenditure. Reduced physical activity is common in the elderly; this is often associated with the abnormality of neurotransmitter transduction as well as reduction of muscle mass/function44. Interestingly, several recent studies have demonstrated that ghrelin administration rescues skeletal muscle atrophy in rodents45.
Under normal physiological conditions, dopamine active transporter (DAT) regulates neurotransmitter dopamine tone by increasing re-uptake of dopamine from synaptic cleft into presynaptic ending of neurons to regulate physical activity46. In obesity-prone rats, total DAT expression and function are significantly reduced compared to that of obesity-resistant rats; this suggests that the DAT function is critical in preventing diet-induced obesity47. Moreover, the loss of dopaminergic neurons in the substantia nigra (SN) is one of the underlying mechanisms for the development of Parkinson’s disease48. Physical activity consists of spontaneous and voluntary activities; spontaneous activity is the term used for obligatory activities including eating and grooming, while voluntary activity is referred to as self-motivated movements such as exercise. Neuronal GHS-R deletion simultaneously promotes both spontaneous and voluntary activity, and has differential effects in different dopaminergic neurons in midbrain9. GHS-R deletion decreases dopaminergic activity in the ventral tegmental area (VTA) by down-regulating DAT, but increases dopaminergic activity by up-regulating the expression of both dopamine synthesis enzyme (TH) and DAT in SN of HFD-fed neuronal-specific GHS-R knockout mice9. The data suggest that dopamine turnover and activity might be elevated in SN, which is in line with increased physical activity observed in HFD-fed neuronal-specific GHS-R knockout mice. We did not observe the enhanced physical activity in our AgRP-neuron-specific GHS-R knockout mice, suggesting that GHS-R in AgRP neurons does not contribute to the physical activity phenotype seen in total neuronal Ghsr knockout mice35. Our data collectively suggest that ghrelin signaling regulates centrally-mediated physical activity through fine-tuning of dopaminergic activity in midbrain.
As summarized in Fig. 1, our studies collectively indicate that neuronal GHS-R is required for the regulation of energy expenditure under obese condition or aging. GHS-R is critical for centrally-mediated thermogenesis and physical activity, specifically, hypothalamic neurons are important sites mediating the effects of GHS-R on thermogenesis, whereas dopaminergic neurons in midbrain control the effects of GHS-R on feeding adaptation and physical activity. Our data also reveal that GHS-R has important role in macrophage polarization; macrophages are key mediator of meta-inflammation in wide range of peripheral tissues, closely linked to insulin resistance. Thus, GHS-R has an important role in pathogenesis of obesity; it is a critical regulator of energy homeostasis, meta-inflammation and metabolic dysfunctions. GHS-R suppression presents a promising strategy for obesity control through multiple cellular and molecular mechanisms, and GHS-R antagonists may serve as unique and powerful drugs for combating obesity, inflammation and insulin resistance.
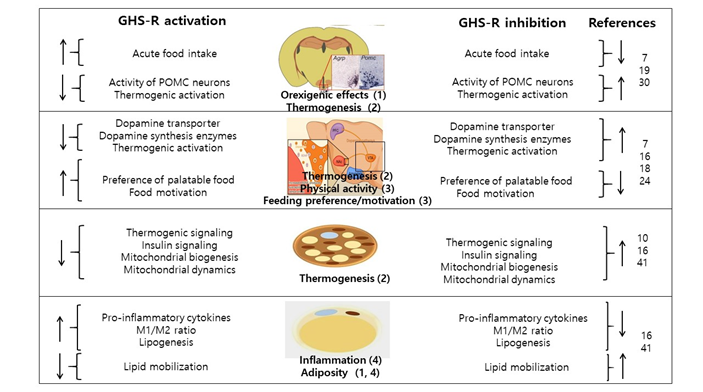
Figure 1. Functions of ghrelin and GHS-R signaling in obesity. Ghrelin and GHS-R signaling under aging or diet-induced obesity: 1) Systemic deletion of GHS-R activates basal firing activity of POMC neurons while GABAergic receptors are blocked, suggesting that ghrelin may activate POMC via an alternative mechanism other than GHS-R. 2) Deletion of GHS-R prevents age-associated thermogenic impairment in BAT by regulating thermogenic and insulin signaling cascades. The neuron-specific deletion of GHS-R increases thermogenesis by augmenting SNS output. GHS-R ablation improves mitochondrial function of brown adipocytes by up-regulating mitochondrial biogenesis and enhancing dynamics. 3) The neuron-specific deletion of GHS-R increases energy expenditure by enhancing physical activity and/or modifying feeding behaviors through modulation of dopaminergic neurons in midbrain. 4) Deletion of GHS-R attenuates inflammation (decreasing M1 macrophages) and reduces adiposity in both white and brown adipose tissues under diet-induced obesity and aging. The number annotated under the image is correlated with the summary points in the legend.
Authorship Contributions
JHL, JD and YS conceived and wrote the manuscript. ES and RCA provided crucial critiques and edited the manuscript.
Acknowledgements
This work was supported by a Basic Science Research Program grant of the National Research Foundation of Korea, the Ministry of Education of Korea (2017R1D1A1B03036210 to JHL), American Heart Association Grant 14GRNT18990019 (YS), and American Diabetes Association Award #1-15-BS-177 (YS). The open access publishing fees for this article have been covered by the Texas A&M University Open Access to Knowledge Fund (OAKFund), supported by the University Libraries and the Office of the Vice President for Research.
References
- Steinberg GR, Schertzer JD. Ampk promotes macrophage fatty acid oxidative metabolism to mitigate inflammation: Implications for diabetes and cardiovascular disease. Immunology and cell biology. 2014; 92: 340-345.
- Kelly B, O'Neill LA. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015; 25: 771-784.
- Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. 2006; 444: 840-846.
- Akhmedov D, Berdeaux R. The effects of obesity on skeletal muscle regeneration. Frontiers in physiology. 2013; 4: 371.
- Cai D, Liu T. Hypothalamic inflammation: A double-edged sword to nutritional diseases. Annals of the New York Academy of Sciences. 2011; 1243: E1-39.
- Cowley MA, Smith RG, Diano S, et al. The distribution and mechanism of action of ghrelin in the cns demonstrates a novel hypothalamic circuit regulating energy homeostasis. Neuron. 2003; 37: 649-661.
- Hill JO, Wyatt HR, Peters JC. Energy balance and obesity. Circulation. 2012; 126: 126-132.
- Kojima M, Kangawa K. Ghrelin: Structure and function. Physiol Rev. 2005; 85: 495-522.
- Lee JH, Lin L, Xu P, et al. Neuronal deletion of ghrelin receptor almost completely prevents diet-induced obesity. Diabetes. 2016; 65: 2169-2178.
- Gnanapavan S, Kola B, Bustin SA, et al. The tissue distribution of the mrna of ghrelin and subtypes of its receptor, ghs-r, in humans. J Clin Endocrinol Metab. 2002; 87: 2988.
- Sun Y, Garcia JM, Smith RG. Ghrelin and growth hormone secretagogue receptor expression in mice during aging. Endocrinology. 2007; 148: 1323-1329.
- Lin L, Lee JH, Bongmba OY, et al. The suppression of ghrelin signaling mitigates age-associated thermogenic impairment. Aging (Albany NY). 2014; 6: 1019-1032.
- Sun Y, Wang P, Zheng H, et al. Ghrelin stimulation of growth hormone release and appetite is mediated through the growth hormone secretagogue receptor. Proc Natl Acad Sci U S A. 2004; 101: 4679-4684.
- Zigman JM, Nakano Y, Coppari R, et al. Mice lacking ghrelin receptors resist the development of diet-induced obesity. J Clin Invest. 2005; 115: 3564-3572.
- Kamegai J, Tamura H, Shimizu T, et al. Chronic central infusion of ghrelin increases hypothalamic neuropeptide y and agouti-related protein mrna levels and body weight in rats. Diabetes. 2001; 50: 2438-2443.
- Morton GJ, Cummings DE, Baskin DG, et al. Central nervous system control of food intake and body weight. Nature. 2006; 443: 289-295.
- Lin L, Saha PK, Ma X, et al. Ablation of ghrelin receptor reduces adiposity and improves insulin sensitivity during aging by regulating fat metabolism in white and brown adipose tissues. Aging Cell. 2011; 10: 996-1010.
- Ma X, Lin L, Yue J, et al. Ghrelin receptor regulates hfcs-induced adipose inflammation and insulin resistance. Nutrition & diabetes. 2013; 3: e99.
- Lin L, Nuotio-Antar AM, Ma X, et al. Ghrelin receptor regulates appetite and satiety during aging in mice by regulating meal frequency and portion size but not total food intake. J Nutr. 2014; 144: 1349-1355.
- Chen SR, Chen H, Zhou JJ, et al. Ghrelin receptors mediate ghrelin-induced excitation of agouti-related protein/neuropeptide y but not pro-opiomelanocortin neurons. Journal of neurochemistry. 2017; 142: 512-520.
- Heppner KM, Piechowski CL, Muller A, et al. Both acyl and des-acyl ghrelin regulate adiposity and glucose metabolism via central nervous system ghrelin receptors. Diabetes. 2014; 63: 122-131.
- Zigman JM, Jones JE, Lee CE, et al. Expression of ghrelin receptor mrna in the rat and the mouse brain. J Comp Neurol. 2006; 494: 528-548.
- Skibicka KP, Hansson C, Alvarez-Crespo M, et al. Ghrelin directly targets the ventral tegmental area to increase food motivation. Neuroscience. 2011; 180: 129-137.
- Abizaid A, Liu ZW, Andrews ZB, et al. Ghrelin modulates the activity and synaptic input organization of midbrain dopamine neurons while promoting appetite. J Clin Invest. 2006; 116: 3229-3239.
- Sirohi S, Van Cleef A, Davis JF. Patterned feeding induces neuroendocrine, behavioral and genetic changes that promote palatable food intake. International journal of obesity. 2017; 41: 412-419.
- Edwards A, Abizaid A. Clarifying the ghrelin system's ability to regulate feeding behaviours despite enigmatic spatial separation of the ghsr and its endogenous ligand. International journal of molecular sciences. 2017; 18.
- Wellman M, Abizaid A. Growth hormone secretagogue receptor dimers: A new pharmacological target. eNeuro. 2015; 2.
- Kern A, Albarran-Zeckler R, Walsh HE, et al. Apo-ghrelin receptor forms heteromers with drd2 in hypothalamic neurons and is essential for anorexigenic effects of drd2 agonism. Neuron. 2012; 73: 317-332.
- Rediger A, Piechowski CL, Yi CX, et al. Mutually opposite signal modulation by hypothalamic heterodimerization of ghrelin and melanocortin-3 receptors. The Journal of biological chemistry. 2011; 286: 39623-39631.
- Scherer PE. Adipose tissue: From lipid storage compartment to endocrine organ. Diabetes. 2006; 55: 1537-1545.
- Cannon B, Nedergaard J. Brown adipose tissue: Function and physiological significance. Physiol Rev. 2004; 84: 277-359.
- Tseng YH, Cypess AM, Kahn CR. Cellular bioenergetics as a target for obesity therapy. Nature reviews. Drug discovery. 2010; 9: 465-482.
- Walden TB, Hansen IR, Timmons JA, et al. Recruited vs. Nonrecruited molecular signatures of brown, "brite," and white adipose tissues. Am J Physiol Endocrinol Metab. 2012; 302: E19-31.
- Contreras C, Nogueiras R, Dieguez C, et al. Traveling from the hypothalamus to the adipose tissue: The thermogenic pathway. Redox biology. 2017; 12: 854-863.
- Wu CS, Bongmba OYN, Yue J, et al. Suppression of ghs-r in agrp neurons mitigates diet-induced obesity by activating thermogenesis. International journal of molecular sciences. 2017; 18.
- Davies LC, Jenkins SJ, Allen JE, et al. Tissue-resident macrophages. Nature immunology. 2013; 14: 986-995.
- Murray RZ, Stow JL. Cytokine secretion in macrophages: Snares, rabs, and membrane trafficking. Frontiers in immunology. 2014; 5: 538.
- Gordon S, Pluddemann A, Martinez Estrada F. Macrophage heterogeneity in tissues: Phenotypic diversity and functions. Immunological reviews. 2014; 262: 36-55.
- Lin L, Lee JH, Buras ED, et al. Ghrelin receptor regulates adipose tissue inflammation in aging. Aging (Albany NY). 2016; 8: 178-191.
- Bulgarelli I, Tamiazzo L, Bresciani E, et al. Desacyl-ghrelin and synthetic gh-secretagogues modulate the production of inflammatory cytokines in mouse microglia cells stimulated by beta-amyloid fibrils. J Neurosci Res. 2009; 87: 2718-2727.
- Avallone R, Demers A, Rodrigue-Way A, et al. A growth hormone-releasing peptide that binds scavenger receptor cd36 and ghrelin receptor up-regulates sterol transporters and cholesterol efflux in macrophages through a peroxisome proliferator-activated receptor gamma-dependent pathway. Mol Endocrinol. 2006; 20: 3165-3178.
- Lucchi C, Costa AM, Giordano C, et al. Involvement of ppargamma in the anticonvulsant activity of ep-80317, a ghrelin receptor antagonist. Frontiers in pharmacology. 2017; 8: 676.
- Ho M, Garnett SP, Baur LA, et al. Impact of dietary and exercise interventions on weight change and metabolic outcomes in obese children and adolescents: A systematic review and meta-analysis of randomized trials. JAMA Pediatr. 2013; 167: 759-768.
- Tieland M, Trouwborst I, Clark BC. Skeletal muscle performance and ageing. Journal of cachexia, sarcopenia and muscle. 2018; 9: 3-19.
- Sjogren M, Duarte AI, McCourt AC, et al. Ghrelin rescues skeletal muscle catabolic profile in the r6/2 mouse model of huntington's disease. Sci Rep. 2017; 7: 13896.
- Bromberg-Martin ES, Matsumoto M, Hikosaka O. Dopamine in motivational control: Rewarding, aversive, and alerting. Neuron. 2010; 68: 815-834.
- Narayanaswami V, Thompson AC, Cassis LA, et al. Diet-induced obesity: Dopamine transporter function, impulsivity and motivation. Int J Obes (Lond). 2013; 37: 1095-1103.
- Dauer W, Przedborski S. Parkinson's disease: Mechanisms and models. Neuron. 2003; 39: 889-909.