
Novel Modality of GSK-3 Inhibition For Treating Neurodegeneration
Ido Rippin, Hagit Eldar-Finkelman*
Department of Human Molecular Genetics and Biochemistry, Sackler School of Medicine, Tel Aviv University, Israel
Neurodegenerative disorders spread over millions of people worldwide and are one of the greatest threats to public health. The prevalence of these disorders is expected to grow exponentially within the next few decades due to the increase in aging population and life span with a resultant enormous social and financial burdens. There is no adequate therapies to these disorders. Therefore, there is an urgent need to accelerate the discovery and development of effective treatments.
Although neurodegenerative disorders are broad ranging and highly complex, they may share overlapping mechanism and common targets for therapeutic interventions. Glycogen synthase kinase-3 (GSK-3) is recognized as an important target in this respect. Since its discovery as a key regulator of glycogen metabolism1, GSK-3 emerged as a multi-tasking kinase involved in multiple cellular processes. In recent years, it became evident that GSK-3 plays key roles in neurological disorders through its ability to alter behavior, cognitive functions, and neuron cell survival2-6. Indeed, many targets controlled by GSK-3 are critically involve in neuron deterioration and disease progression. These include, for example, the microtubule associate protein tau7, pro-inflammatory factors8, heat shock proteins9, brain derived neurotrophic factor BDNF10 and Wnt signaling pathway11. Worth mentioning is our recent findings that linked GSK-3 with impaired cellular clearance through negative regulation of autophagy and lysosomal activity12. Inefficient autophagy is coupled with accumulation and formation of typical neurotoxic aggregates in neurodegeneration conditions13,14. Altogether, this raise the paradigm that GSK-3 inhibition is a potent and beneficial therapeutic for neurodegeneration.
Development and design of specific inhibitors of protein kinases is a focus of many drug discovery programs. Most protein kinase inhibitors developed so far are small molecules that compete with the ATP binding site of the kinase. This type of inhibition, although powerful, often has limited specificity because the ATP binding site is highly conserved among protein kinases15,16. Indeed, clinically approved ATP competitive drugs are noted to carry with them the risks for severe side effects due to specificity issues and for being ineffective against drug resistance mutations17-20. Thus, it was clear to us that a different type of GSK-3 inhibitors that do not bind (exclusively) to its ATP binding site is a favorable choice for clinical practice.
To fulfill this requirement we focused our attention on developing substrate competitive inhibitors (SCIs) for GSK-3. These compounds are short peptides that mimic substrate sequences and bind to the substrate-binding cavity of the kinase21. SCIs are considered specific because the substrate binding site is less conserved among the protein kinase family15,16, in addition, they are expected to be less prone to drug-induced resistance due to the their large binding surface. We further showed that it is possible to optimize the potency of GSK-3 SCIs by strengthening their interactions with the GSK-3-substrates binding site using computational and biological approach22. The first generation of GSK-3 SCIs function as pseudo- substrates23, namely, they are ‘phosphorylated invalid’ substrates24 (Fig. 1). Recently, we discovered a new type of SCI termed ‘substrate converted into an inhibitor’25. The discovery was unexpected as we found that when we turn the pseudosubstrate back to a substrate it functions as an inhibitor. Namely, the inhibitor is a ‘real’ substrate, it binds to GSK-3, and upon phosphorylation (by the kinase) it turns in-situ into a potent inhibitor (Fig. 1). Thus, in contrast to ‘normal’ substrates that will immediately dissociate from the enzyme after being phosphorylated, phosphorylation of the ‘substrate converted into an inhibitor’ will result in a tighter binding to the kinase25. In fact, the ‘substrate converted into an inhibitor’ differs from the original sequence composition of the natural substrate in at least two positions. These simultaneous changes are key for its accompanied behavior as an inhibitor.
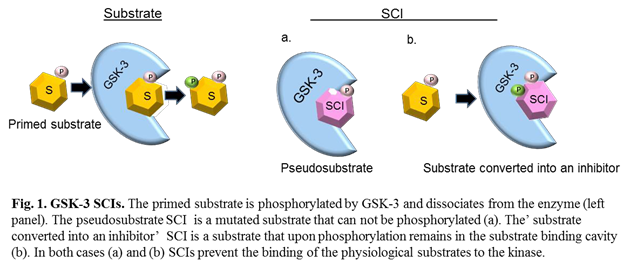
The use of GSK-3 SCIs indeed provided proof of concept. Treatment with GSK-3 SCIs improved Alzheimer’s brain pathology and reversed cognitive decline12,25. They improved clinical symptoms in multiple sclerosis mouse model26, and enhanced cognitive skills in FMRP-/- mice that represent Fragile X syndrome27.
There are several advantages of the new modality of ‘substrate converted into an inhibitor’. First, the strict requirement for being phosphorylated increases specificity, thus, limiting off target effects, Second, the inhibitor is effective only toward active GSK-3 sparing the inactive GSK-3 population ‘unharmed’ by the inhibitor. Third, the design of this type of inhibitor may be applied to many other protein kinases.
In summary, inhibition of GSK-3 has been considered a promising therapeutic approach for treating several neurodegenerative disorders including Alzheimer’s and Parkinson’s disease, depressive behavior, autism, and Huntington’s disease. However, none of the GSK-3 inhibitors that were developed reached the market. Our suggestion is that ‘correct’ inhibition of GSK-3 is the key for a successful use of such inhibitors. Hence, the different inhibition modality based on substrate competition and, in particular, the type of ‘substrate converted into an inhibitor’ described here is a potential approach for fruitful treatment in the clinic.
References
- Woodgett JR, Cohen P. Multisite phosphorylation of glycogen synthase. Molecular basis for the substrate specificity of glycogen synthase kinase-3 and casein kinase- II (glycogen synthase kinase-5). Biochim Biophys Acta. 1984; 788: 339-347.
- Beurel E, Grieco SF, Jope RS. Glycogen synthase kinase-3 (GSK3): regulation, actions, and diseases. Pharmacol Ther. 2015; 148: 114-131.
- Hernandez F, Lucas JJ, Avila J. GSK3 and tau: two convergence points in Alzheimer's disease. J Alzheimers Dis. 2013; 33 Suppl 1: S141-144.
- Eldar-Finkelman H, Martinez A. GSK-3 Inhibitors: Preclinical and Clinical Focus on CNS. Front Mol Neurosci. 2011; 4: 32.
- Patel P, Woodgett JR. Glycogen Synthase Kinase 3: A Kinase for All Pathways. Curr Top Dev Biol. 2017; 123: 277-302.
- Lovestone S, Boada M, Dubois B, et al. A phase II trial of tideglusib in Alzheimer's disease. J Alzheimers Dis. 2015; 45: 75-88.
- Hanger DP, Anderton BH, Noble W. Tau phosphorylation: the therapeutic challenge for neurodegenerative disease. Trends Mol Med. 2009; 15: 112-119.
- Jope RS, Yuskaitis CJ, Beurel E. Glycogen synthase kinase-3 (GSK3): inflammation, diseases, and therapeutics. Neurochem. Res. 2007; 32: 577-595.
- Chu B, Soncin F, Price BD, et al. Sequential phosphorylation by mitogen-activated protein kinase and glycogen synthase kinase 3 represses transcriptional activation by heat shock factor-1. J Biol Chem. 1996; 271: 30847-30857.
- Shaltiel G, Chen G, Manji HK. Neurotrophic signaling cascades in the pathophysiology and treatment of bipolar disorder. Curr Opin Pharmacol. 2007; 7: 22-26.
- O'Brien WT, Klein, PS. Validating GSK3 as an in vivo target of lithium action. Biochem Soc Trans. 2009; 37: 1133-1138.
- Avrahami L, Farfara D, Shaham-Kol M, et al. Inhibition of GSK-3 Ameliorates beta-Amyloid (A-beta) Pathology and Restores Lysosomal Acidification and mTOR Activity in the Alzheimer's Disease Mouse Model. In vivo and In vitro Studies. J Biol Chem. 2013; 288: 1295-1306.
- Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med. 2013; 19: 983-997.
- Rubinsztein DC, Codogno P, Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov. 2012; 11: 709-730.
- Hanks SK, Quinn AM, Hunter T. The protein kinase family: conserved features and deduced phylogeny of the catalytic domains. Science. 1988; 241: 42-52.
- Taylor SS, Radzio-Andzelm E, Hunter T. How do protein kinases discriminate between serine/threonine and tyrosine? Structural insights from the insulin receptor protein-tyrosine kinase. FASEB J. 1995; 9: 1255-1266.
- Bain J1, Plater L, Elliott M, et al. (2007) The selectivity of protein kinase inhibitors: a further update. Biochem. J. 408, 297-315.
- Dar AC, Shokat KM. The evolution of protein kinase inhibitors from antagonists to agonists of cellular signaling. Annu Rev Biochem. 2011; 80: 769-795.
- Rosti G, Castagnetti F1, Gugliotta G, et al. Tyrosine kinase inhibitors in chronic myeloid leukaemia: which, when, for whom. Nat Rev Clin Oncol. 2017; 14: 141-154.
- Dong L, Lei D, Zhang H. Clinical strategies for acquired epidermal growth factor receptor tyrosine kinase inhibitor resistance in non-small-cell lung cancer patients. Oncotarget. 2017; 8: 64600-64606.
- Avrahami L1, Licht-Murava A, Eisenstein M, et al. GSK-3 inhibition: Achieving moderate efficacy with high selectivity. Biochim Biophys Acta. 2013.
- Licht-Murava A, Plotkin B, Eisenstein M, et al. Elucidating substrate and inhibitor binding sites on the surface of GSK-3beta and the refinement of a competitive inhibitor. J Mol Biol. 2011; 408: 366-378.
- Kemp BE, Pearson RB, House CM. Pseudosubstrate-based peptide inhibitors. Methods Enzymol. 1991; 201: 287-304.
- Plotkin B, Kaidanovich O, Talior I, et al. Insulin mimetic action of synthetic phosphorylated peptide inhibitors of glycogen synthase kinase-3. J Pharmacol Exp Ther. 2003; 974-980.
- Licht-Murava A, Paz R, Vaks L, et al. A unique type of GSK-3 inhibitor brings new opportunities to the clinic. Science Signaling. 2016; 9: ra110.
- Beurel E, Kaidanovich-Beilin O, Yeh WI, et al. Regulation of Th1 cells and experimental autoimmune encephalomyelitis by glycogen synthase kinase-3. J Immunol. 2013; 190: 5000-5011.
- Pardo M, Cheng Y, Velmeshev D, et al. Intranasal siRNA administration reveals IGF2 deficiency contributes to impaired cognition in Fragile X syndrome mice. JCI Insight. 2017; 2: e91782.