
Neuropathies of Stuve-Wiedemann Syndrome due to mutations in leukemia inhibitory factor receptor (LIFR) gene
Alexandra E. Oxford, Cheryl L. Jorcyk, Julia Thom Oxford*
Abstract
Stüve-Wiedemann syndrome (STWS; OMIM #610559) is a rare disease that results in dysfunction of the autonomic nervous system, which controls involuntary processes such as breathing rate and body temperature. In infants, this can result in respiratory distress, feeding and swallowing difficulties, and hyperthermic episodes. Individuals may sweat excessively when body temperature is not elevated. Additionally, individuals have reduced ability to feel pain and may lose reflexes such as the corneal reflex that normally causes one to blink, and the patellar reflex resulting in the knee-jerk. STWS usually results in infant mortality, yet some STWS patients survive into early adulthood. STWS is caused by a mutation in the leukemia inhibitory factor receptor (LIFR) gene, which is inherited in an autosomal-recessive pattern. Most LIFR mutations resulting in STWS cause instability of the mRNA due to frameshift mutations leading to premature stop codons, which prevent the formation of LIFR protein. STWS is managed on a symptomatic basis as no treatment is currently available.
Introduction
Stüve-Wiedemann syndrome (STWS) was first described in 1971 in two sisters with congenital bowing of the tibia and femur. While both patients experienced respiratory distress and died within a few days, one also suffered from hyperthermia1,2. Although the prognosis of STWS remains poor today, recent reports show that some STWS patients survive into adulthood3,4. STWS patients are phenotypically identical to Schwartz-Jampel syndrome type 2 (SJS2) patients5,6, and both SJS2 and STWS have been linked to a mutation of the leukemia inhibitory factor receptor (LIFR) gene on chromosome 5p13.1, and SJS2 and STWS are now considered the same syndrome7. STWS is found across multiple ethnic groups in multiple regions of the world including North America, Europe, and the Middle East7,8-12.
The skeletal features of STWS place it within the bent-bone dysplasias13, characterized by bowing of the long bones with cortical thickening and rarefaction, wide and blurred margins of the metaphyses, contracture and limited mobility of elbows and knees, osteopenia, flared iliac wings, hypoplasia of the lower ilia, and an abnormal trabecular shape1,7,9,14. Progressive bowing of the long bones, severe spinal deformities, osteoporosis and spontaneous fractures occur. Joints may be prominent with restricted mobility. While skeletal features are important, skeletal symptoms will not be the focus of this review, and we will restrict our discussion to nervous system symptoms.
Clinical manifestations
Many individuals with STWS do not survive beyond the first few months of life due to respiratory distress, difficulties with feeding and swallowing, and hyperthermic episodes1,7-9,15-17. Patients that do survive show an improvement in prognosis as a normal breathing rhythm is established and the ability to swallow is gained, yet difficulties with swallowing can still occur later in childhood 9. Temperature instability is accompanied by excessive and paradoxical sweating. Other signs of dysautonomia persist, such as a smooth tongue, absent corneal and patellar reflexes, and reduced pain sensation. Physical growth and motor development is delayed, but intelligence is normal7-9. The smooth tongue phenotype seen in STWS is due to a lack of fungiform papillae, normally found on the upper surface of the tongue. The inner structural connective tissue of fungiform papillae is highly innervated and provides the ability to distinguish taste. Taste bud development and maintenance is strictly dependent on the innervation during early postnatal development, and fungiform papillae do not develop when innervation is obstructed18. Additionally, fungiform papillae do not develop in the absence of a functional LIFR or signaling by LIF and CNTF19. Lack of fungiform papillae is both a structural and a neurological pathology of STWS. A smooth tongue due to their absence is a key indicator of the role LIFR plays in normal neuronal development of taste buds, and an important symptom that STWS patients encounter.
References
The mutated LIFR gene is inherited in an autosomal recessive pattern. The mutations reported to date have been missense or nonsense mutations, with the majority of mutations occurring within the exons encoding the extracellular domain7. The LIFR gene has 20 exons, 19 of which encode amino acids of this transmembrane protein which is 1,097 amino acids long7,20,21.
Mouse genetic models have informed us of phenotypes resembling STWS. LIFR null mice display a strikingly similar phenotype consistent with STWS patients22. The number of motor neurons in the facial nucleus, lumbar spinal cord, and nucleus ambiguous are severely reduced in mice that do not express LIFR23. Importantly for STWS, the nucleus ambiguus innervates the esophagus, pharynx, larynx, and coordinates swallowing and is responsible for initiating the respiratory rhythm.
Genetic Variability
Although not all STWS patients have an identified LIFR mutation9,17, other genes responsible for the STWS phenotype have not been identified. Additional genes within the chromosomal region 5p13.1 from locus D5S194 to D5S1457 mapped by Dagoneau et al. included FLJ39155 (EGFLAM or Pikachurin, a proteoglycan), disabled-2 (DAB2), complement 9 (C9), Fyn-binding protein (FYB), and oncostatin M receptor (OSMR)7. While these genes may play a role as disease modifiers or may be potential candidates for those cases of STWS that are not linked to a mutation in LIFR, to date, limited information exists to support the likelihood of the involvement of these genes in skeletal, respiratory or neurological symptoms associated with the syndrome. Respiratory function has been associated with DAB-2. DAB-2 is a clathrin-associated sorting protein (CLASP) that contributes to clathrin recruitment, vesicle formation, and cargo selection. In the lungs, cystic fibrosis transmembrane conductance regulator, a cAMP-activated chloride channel expressed in the apical plasma membrane of human airway epithelial cells, is endocytosed in a DAB-2-dependent manner24. DAB-2 also plays a role in bone morphogenetic protein signaling25 and nerve cell differentiation26. Additionally, a neurologic function has been described for FYB through an association with hereditary motor-sensory neuropathy27. OSMR, like LIFR, is able to bind to glycoprotein (gp)130 to form heterodimers within the plasma membrane. OSMR has also been implicated in the respiratory system28, and the nervous system29, as well as metabolic symptoms such as mature-onset obesity, severe hepatic steatosis, and insulin resistance30.
OSMR plays a key role in bone homeostasis as demonstrated by the OSMR(-/-) mouse, which exhibits an osteopetrotic phenotype due to an effect on osteoclast differentiation31,32. The OSMR (-/-) mouse however, does not display the autonomic manifestations seen in STWS patients, suggesting that loss of OSMR by itself would not result in STWS. Therefore, the presence of OSMR within the candidate region is likely due to gene duplication and its presence within the candidate region is probably coincidence.
Proteins that play an essential role in the LIFR signaling pathway as ligands or as competing receptor, such as the cytokine receptor-like factor 1 (CRLF1), cardiotrophin-like cytokine factor 1 (CLCF1), neuropoietin (NP), and ciliary neurotrophic factor (CNTF) may play a role in the neuronal symptoms of STWS9 and are discussed in more detail.
The lifr protein and signalingReferences
The LIFR Protein
The LIFR protein (glycoprotein-190; gp 190) is composed of a signal peptide followed by three main domains. The extracellular domain (45-833aa) includes two cytokine receptor homology domains (CRH1 and CRH 2), one Ig-like domain (Ig), and one type III fibronectin domain with three modules (FNIII). The transmembrane domain (TM; 834-858aa) is located between the extracellular domain and the cytoplasmic domain (CD; 859-1097aa) (Figure 1).
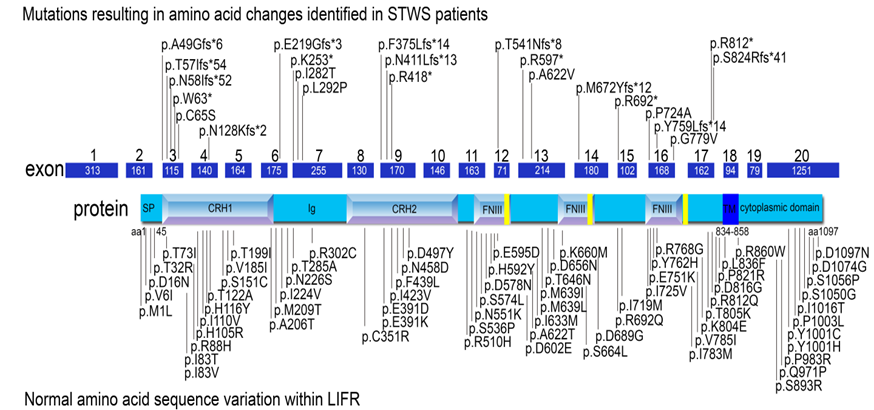
Figure 1. Known mutations in LIFR and normal sequence variants of LIFR. The LIFR protein is encoded by 19 exons within a 20 exon mRNA. The domains are illustrated within their corresponding region of the protein. SP: signal peptide; CRH1 and CRH2: cytokine receptor homology domains 1 and 2, Ig: Immunoglobulin-like domain; FNIII: fibronectin type III domain (433-530; 541-623; 729-821); TM: transmembrane domain (834-858); CD: cytoplasmic domain. Mutations observed in STWS patients are illustrated above the gene model86 and the normal variations within the LIFR gene are indicated below the gene model87. Currently, no pattern has emerged for the symptoms experienced by STWS patients and their specific mutation.
The LIFR Signaling Pathway
LIFR binds with low affinity to several IL-6 cytokine family members, including leukemia inhibitory factor (LIF), oncostatin-M (OSM), cardiotrophin-1 (CT-1, also abbreviated as CTF-1), ciliary neurotrophic factor (CNTF), neuropoietin (NP also abbreviated CTF2 in mouse, and CTF2P in human), and cardiotrophin-like cytokine factor 1 (CLCF-1, also abbreviated as CLC)33-36 (Figure 2). Both LIF and OSM bind to the LIFR. LIF binds to LIFRβ, which then recruits gp130 for higher affinity and cell signaling21. In contrast, OSM binds gp130 with low affinity but has little to no biological activity unless a second receptor chain is recruited, either the LIFRβ or the more highly specific OSMRβ37-40. CT-1 binds to gp130 and LIFR41, while CNTF first binds to ciliary neurotrophic factor receptor (CNTFRα) before recruiting LIFR42. NP signals through a receptor complex comprising CNTFRα, gp130, and LIFR43. Cardiotrophin-like cytokine factor 1 (CLCF-1) forms a heterodimer with either cytokine receptor-like factor 1 (CRLF1) or soluble ciliary neurotrophic factor receptor (sCNTFR) and competes for this same receptor complex9,44-46. CNTF, LIF, and NP may be responsible for neuronal development and survival, as CTNF possesses neurotrophic activity and can enhance precursor self-renewal and expansion of neuronal stem cells47,48. CTNF and LIF may play a role in motor neuron innervation49-51and NP can sustain in vitro survival of embryonic motor neurons and can increase the proliferation of neural precursors52 (Table 1).
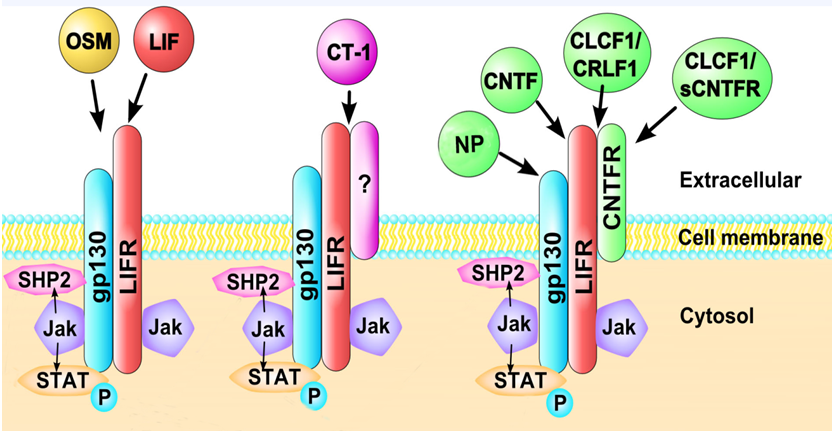
Figure 2. Cytokines interacting with LIFR.Leukemia inhibitory factor (LIF), oncostatin-M (OSM), cardiotrophin-1 (CT-1), ciliary neurotrophic factor (CNTF), neuropoietin (NP), and cardiotrophin-like cytokine factor 1 (CLCF-1/CRLF1 or a soluble form sCNTRF) bind to LIFR-containing heterodimeric receptors. LIFR can associate with other receptor subunits including gp130 and CNTFR. OSM and LIF interact with LIFR in association with gp130. CT-1 associates with LIFR with gp130 and potentially other receptor subunits. CNTF interacts with LIFR in association with gp130 and CNTFR, as does CLCF-1/CRLF1, NP and sCRLF1. Additionally, binding may occur in a competitive fashion with other cytokines, increasing the complexity of signaling through LIFR.
| STWS phenotype | Cytokine |
|---|---|
| Smooth tongue | LIF, CNTF |
| Osteopenia | LIF, OSM, CT-1 |
| Cardiovascular malfunctions | LIF, CT-1 |
| Paradoxical sweating | CLCF1/CRLF1 |
| Dysphagia | CT-1?, OSM?, CLCF1/CRLF1? |
| Respiratory distress | CT-1?, OSM? |
| Short stature | CNTF |
Table 1.LIFR ligand and associated symptoms
Each of the cytokines signal through the leukemia inhibitory factor receptor (LIFR). In STWS, the LIFR gene is mutated, and due to the mutation, cytokine signaling is blocked, and the corresponding manifestations of STWS occur. Only symptoms for which the mechanism has been explored are listed. Those followed by a question mark are still under considerable speculation. LIF: leukemia inhibitory factor; CNTF: ciliary neurotrophic factor; OSM: oncostatin-M; CT-1: cardiotrophin-1; CLCF1: cardiotrophin-like cytokine factor-1; CRLF1: cytokine receptor-like factor-1. Neuropoietin is not included in the table.
Lifr ligands linked to neuronal development
Leukemia Inhibitory Factor
LIF is a pleiotropic cytokine, secreted by a variety of cell types, including epithelial and stromal cells53, osteoblasts54,55, bone marrow stromal cells, fibroblasts, astrocytes, heart myoblasts, T lymphocytes, monocytes, and thymic epithelial cells among others56.
stify"
LIF acts as a neurotrophic factor57 and may play a role in motor neuron innervation49-51. LIF is known to stimulate cholinergic differentiation in sympathetic neurons, inducing choline acetyltransferase gene expression, which in turn promotes the survival of cholinergic neurons58. Similarly, LIF is important for the cholinergic transdifferentiation of cardiac sympathetic neurons. Hence, it is likely that a lack of LIF downstream signaling leads to the cardiovascular phenotype seen in STWS59-61. LIF also has neuromodulary roles in the respiratory airways62. LIF enhances the differentiation and survival of motor neurons.
Leukemia Inhibitory Factor
OSM shares similarities with LIF, as both are able to induce the differentiation of myeloid leukemia cells to macrophage-like cells in mice63. LIF and OSM are located near each other on chromosome 22, and their arrangements suggest that LIF and OSM may be the result of a gene duplication event of an ancestral gene64.
The reduction in motor neurons seen in STWS is due in part to the inability of OSM to signal through the mutated LIFR23. OSM is likely to be one of the LIFR ligands responsible for the respiratory distress and dysphagia seen in STWS.
Cardiotrophin-1 (CT-1)
CT-1 plays an important role in the cholinergic transdifferentiation of cardiac sympathetic neurons in rodents59,65. Specifically, CT-1 loss reduced the number of preganglionic sympathetic neurons, which are important for inducing heart rate, ventricular pressure, and contractility66. CT-1 is also known to induce cardiac myocyte hypertrophy and vascular smooth muscle cell proliferation in vitro60. Therefore, the lack of CT-1 signaling in STWS may play a role in the cardiovascular phenotype67 and in motor neuron survival68. A role for CT-1 in airway smooth muscle cells has been identified69,70, and therefore, an absence of CT-1 signaling in STWS may contribute to the respiratory phenotype of STWS. Similarly, CT-1 is important for the cholinergic transdifferentiation of cardiac sympathetic neurons. Hence, it is likely that a lack of CT-1 downstream signaling leads to the cardiovascular phenotype seen in STWS59-61. The reduction in motor neurons seen in STWS is due in part to the inability of CT-1 to signal through the mutated LIFR23. CT-1 is likely to be responsible for the respiratory distress and dysphagia seen in STWS.
Ciliary Neurotrophic Factor (CNTF)
STWS includes autonomic nerve dysfunction3, and ciliary neurotrophic factor receptor (CNTFR) gene has also been linked to autonomic nervous system dysfunction71,72. CNTF is expressed in the developing nervous system, particularly in the motor neurons, and it plays a role in motor neuron survival23. CNTF are known to stimulate cholinergic differentiation in sympathetic neurons, inducing choline acetyltransferase gene expression, which in turn promotes the survival of cholinergic neurons58. CNTF enhances the differentiation and survival of motor neurons. Mice deficient in CNTF do not show a decrease in motor neurons.
Neuropoietin (NP; CTF2; CTF2P)
Neuropoietin is encoded by the CTF2 gene in the mouse and CTF2P in human. The approved name is cardiotrophin 2, pseudogene in humans. It was described in 2004 by Derouet and colleagues52, located on mouse chromosome 7 (choromosome 16 in humans) in close proximity to the cardiotrophin-1 gene. NP or CTF2, is predominantly expressed in mouse neuroepithelia during embryonic life, acts through a receptor complex comprising CNTFRα component, gp 130, and LIF receptor. Like CNTF, it promotes the survival of embryonic motor neurons and could increase the proliferation of neural precursor cells. Interestingly, the human gene has evolved into a pseudogene due to an 8 base pair deletion causing a disruption in the reading frame. This suggests that signaling via CNTF can compensate in the absence of a functional CTF2 in humans. While NP (CTF2) plays key roles in other mammals, it does not play a role in the symptoms of STWS.
NP is highly expressed in embryonic neuroepithelia and in the retina. NP can sustain the in vitro survival of embryonic motor neurons and could increase the proliferation of neural precursors. NP induces neuroepithelial cells to differentiate into astrocytes, and does so in coordination with bone morphogenetic protein 2 (BMP2)43,52,73.
Cardiotrophin-like Cytokine Factor-1 (CLCF-1)
Crisponi syndrome and cold-induced sweating syndrome share some features with STWS, such as feeding difficulties, trismus, paradoxical sweating (i.e., sweating with low body temperatures72), and hyperthermic episodes74. Crisponi syndrome is now considered to be the same disorder as cold-induced sweating syndrome75, and cold-induced sweating syndrome is caused by mutations in the CLCF-1 or CRLF-1 genes. CLCF-1 binds with either CRLF-1 (cytokine receptor-like factor-1) or sCNTFR (soluble ciliary neurotrophic factor receptor) and then competes with CNTF (ciliary neurotrophic factor) for the receptor complex composed of CNTFR, LIFR and gp13043,45,76. Therefore, it is likely that the dysautonomic symptoms seen in STWS are caused by a lack of CLCF-1/CRLF-1 signaling due to a mutated LIFR gene. Additionally, Clcf-1/Crlf-1 is responsible for cholinergic differentiation of neurons innervating sweat glands77. Mice lacking Crlf-1, Cntfr and Clcf-1 are unable to suckle and die shortly after birth78. These mice also have a reduced number of motor neurons in the facial nucleus79. Therefore, a lack of CLCF-1/CRLF-1 signaling leads to dysphagia and facial muscle contractions observed in STWS.
Future directions
Although most reported cases of STWS are associated with a mutation in the LIFR gene, some diagnosed cases do not have a demonstrable mutation in the LIFR gene. Although it is possible that those patients were misdiagnosed, as STWS shares many traits with other syndromes, it is also very likely that other genes play a role in STWS. Identical frameshift mutations in the LIFR gene in different individuals have also been reported to show differing outcomes or severity.
Even in the cases where LIFR signaling is known to be the root cause of STWS, connections between signaling and symptoms have not been fully elucidated. As many cytokines possess redundant roles, it is likely that there is overlap in function. Additional proteins involved in STWS will likely be discovered as additional research is carried out.
The future holds promise for potential new treatments for STWS. In the case where a single mutation causes STWS, gene editing technologies such as CRISPR/Cas9 may be used to directly modify and correct the STWS associated change in the genome80. If successful, correcting the LIFR gene could result in the expression of a normal and functional LIFR protein. Antisense mediated exon skipping may also be a useful strategy in cases where exons encode independently folding domains within the protein or where the remaining domain can still fold stably and correctly and carry out the function of the normal protein81. Exon skipping strategies may be especially applicable in cases where a mutation causes a change in reading frame, and skipping of an exon will restore the proper reading frame. Many of the identified disease causing mutations are nonsense mutations, introducing a premature stop codon within the coding regions, resulting in the production of a truncated protein or alternatively, mRNA template degradation. It may be possible to promote read-through of premature stop codons using aminoglycoside antibiotic treatment82-84 that influences the fidelity of the stop codon recognition by changing the conformation of the rRNA. 5-(fluorophenyl)-1,2,4-oxadiazolyl-benzoic acid has also been suggested to suppress nonsense mutations by read-through and is being considered for the treatment of cystic fibrosis and may also be applicable for STWS nonsense mutations85.
Conclusions
STWS is a rare bent-bone dysplasia with dysautonomic manifestations that is generally caused by the autosomal recessive inheritance of a mutated LIFR gene. The symptoms of STWS are the result of a lack of LIFR signaling, although the exact mechanisms remain unclear for most phenomena. There is currently no treatment available for STWS. Instead, symptoms are managed accordingly. The prognosis remains poor and there are many unanswered questions regarding its pathology. Therefore, further research is needed to provide a better mechanistic understanding as well as to make progress toward novel therapies that take advantage of what we do know about the targeted manipulation of specific signaling pathways.
Acknowledgement
Authors acknowledge support by Institutional Development Award (IDeA) Program from the National Institute of General Medical Sciences of the National Institutes of Health under Grants #P20GM103408 and P20GM109095. We also acknowledge support from The Biomolecular Research Center at Boise State, the MJ Murdock Charitable Trust; Lori and Duane Steuckle, and the Idaho State Board of Education.
References
- Stuve A, Wiedemann HR. Congenital bowing of the long bones in two sisters. Lancet. 1971; 2:495.
- Wiedemann H, Stueve A. Stüve?Wiedemann syndrome: Update and historical footnote. Am J Med Genet. 1996; 63:12–16.
- Rocco M Di, Stella G. Long?term survival in Stuve?Wiedemann syndrome: A neuro?myo?skeletal disorder with manifestations of dysautonomia. Am J Med Genet. 200; 118A:362–368.
- Pizones J, Sponseller PD, Izquirdo E, Sanz E, Sanchez-Martinez F, Alvarez P, et al. Delayed tetraplegia after thoracolumnbar scoliosis surgery in Stuve-Wiedemann syndrome. Spine Deform. 2013; 1:72–78.
- Chen E, Cotter P. Characterization of a long?term survivor with Stüve?Wiedemann syndrome and mosaicism of a supernumerary marker chromosome. Am J Med Genet. 2001; 101:240–245.
- Superti?Furga A, Tenconi R. Schwartz?Jampel syndrome type 2 and Stüve?Wiedemann syndrome: A case for “Lumping.” Am J Med Genet. 1998; 78:150–154.
- Dagoneau N, Scheffer D, Huber C, Al-Gazali LI, Di Rocco M, Godard A, et al. Null Leukemia Inhibitory Factor Receptor (LIFR) Mutations in Stüve-Wiedemann/Schwartz-Jampel Type 2 Syndrome. Am J Hum Genet. 2004; 74: 298–305.
- Al-Gazali LI, Ravenscroft A, Feng A, Shubbar A, Al-Saggaf A, Haas D. Stüve-Wiedemann syndrome in children surviving infancy: clinical and radiological features. Clin Dysmorphol. 2003; 12:1–8.
- Jung C, Dagoneau N, Baujat G, Merrer M, David A, Rocco M, et al. Stuve-Wiedemann syndrome: long-term follow-up and genetic heterogeneity. Clin Genet. 2010; 77:266–72.
- Al-Gazali L, Ali BR. Mutations of a country: a mutation review of single gene disorders in the United Arab Emirates (UAE). Hum Mutat. 2010; 31:505–520.
- Koul R, Al-Kindy A, Mani R, Sankhla D, Al-Futaisi A. One in three: congenital bent bone disease and intermittent hyperthermia in three siblings with stuve-wiedemann syndrome. Sultan Qaboos Univ Med J. 2013; 13:301–5.
- Knipe M, Stanbury R, Unger S, Chakraborty M. Stuve-Wiedemann syndrome with a novel mutation. BMJ Case Reports. 2015; doi: 10.1136/brc-2015-212032.
- Warman ML, Cormier-Daire V, Hall C, Krakow D, Lachman R, LeMerrer M, et al. Nosology and classification of genetic skeletal disorders: 2010 revision. Am J Med Genet A. 2011; 155A:943–968.
- Cormier-Daire V, Munnich A, Lyonnet S, Rustin P, Delezoide AL, Maroteaux P, et al. Presentation of six cases of Stüve-Wiedemann syndrome. Pediatr Radiol 1998; 28:776–780.
- Bonthuis D, Morava E. Stuve Wiedemann syndrome and related syndromes: case report and possible anesthetic complications. Paediatr Anesth. 2009; 19:212–217.
- Corona-Rivera JR, Cormier-Daire V, Dagoneau N, Coello-Ramírez P, López-Marure E, Romo-Huerta CO, et al. Abnormal oral-pharyngeal swallowing as cause of morbidity and early death in Stüve-Wiedemann syndrome. Eur J Med Genet. 2009; 52:242–6.
- Kaissi A, Rumpler M, Csepan R, Grill F, Klaushofer K. Congenital contractures and distinctive phenotypic features consistent with Stuve-Wiedmann syndrome in a male infant. Cases J. 2008; 1:121.
- Sollars SI, Bernstein IL. Neonatal chorda tympani transection permanently disrupts fungiform taste bud and papilla structure in the rat. Physiol Behav. 2000; 69:439-44.
- Gardiner J, Barton D, Vanslambrouck JM, Braet F, Hall D, Marc J, et al. Defects in tongue papillae and taste sensation indicate a problem with neurotrophic support in various neurological diseases. Neuroscientist. 2008; 14:240–250.
- Tomida M, Gotoh O. Structure of the gene encoding the human differentiation-stimulating factor/leukemia inhibitory factor receptor. J Biochem. 1996; 120:201–5.
- Gearing DP, Thut CJ, VandeBos T, Gimpel SD, Delaney PB, King J, et al. Leukemia inhibitory factor receptor is structurally related to the IL-6 signal transducer, gp130. EMBO J 1991; 10:2839–48.
- Ware CB, Horowitz MC, Renshaw BR, Hunt JS, Liggitt D, Koblar SA, et al. Targeted disruption of the low-affinity leukemia inhibitory factor receptor gene causes placental, skeletal, neural and metabolic defects and results in perinatal death. Development. 1995; 121:1283-99.
- Li M, Sendtner M, Smith A. Essential function of LIF receptor in motor neurons. Nature. 1995; 378:724–727.
- Cihil KM, Ellinger P, Fellows A, Stolz DB, Madden DR, Swiatecka-Urban A. Disabled-2 protein facilitates assembly polypeptide-2-independent recruitment of cystic fibrosis transmembrane conductance regulator to endocytic vesicles in polarized human airway epithelial cells. J Biol Chem. 2012; 287:15087–99.
- Kim J-D, Kang H, Larrivée B, Lee MY, Mettlen M, Schmid SL, et al. Context-dependent proangiogenic function of bone morphogenetic protein signaling is mediated by disabled homolog 2. Dev Cell. 2012; 23:441–8.
- Huang C-H, Cheng J-C, Chen J-C, Tseng C-P. Evaluation of the role of Disabled-2 in nerve growth factor-mediated neurite outgrowth and cellular signalling. Cell Signal. 2007; 19:1339–47.
- Nevsímalová S, Prazák J, Herzog P, Seemanová E. Duffy locus linkage and HLA antigens in hereditary motor-sensory neuropathy. Schweiz Arch Neurol Psychiatr. 1991; 142:19–29.
- Chen D, Chu C-Y, Chen C-Y, Yang H-C, Chiang Y-Y, Lin T-Y, et al. Expression of short-form oncostatin M receptor as a decoy receptor in lung adenocarcinomas. J Pathol. 2008; 215:290–9.
- Morikawa Y: Oncostatin M in the development of the nervous system. Anat Sci Int 2005, 80:53–9.
- Komori T, Tanaka M, Senba E, Miyajima A, Morikawa Y. Lack of oncostatin M receptor β leads to adipose tissue inflammation and insulin resistance by switching macrophage phenotype. J Biol Chem. 2013; 288:21861–75.
- Tanaka M, Hirabayashi Y, Sekiguchi T, Inoue T, Katsuki M, Miyajima A. Targeted disruption of oncostatin M receptor results in altered hematopoiesis. Blood 2003; 102:3154–62.
- Walker EC, McGregor NE, Poulton IJ, Solano M, Pompolo S, Fernandes TJ, et al. Oncostatin M promotes bone formation independently of resorption when signaling through leukemia inhibitory factor receptor in mice. J Clin Invest. 2010; 120:582–92.
- Gough NM, Gearing DP, King JA, Willson TA, Hilton DJ, Nicola NA, et al. Molecular cloning and expression of the human homologue of the murine gene encoding myeloid leukemia-inhibitory factor. Proc Natl Acad Sci U S A. 1988; 85:2623–7.
- Malik N, Kallestad JC, Gunderson NL, Austin SD, Neubauer MG, Ochs V, et al. Molecular cloning, sequence analysis, and functional expression of a novel growth regulator, oncostatin M. Mol Cell Biol. 1989; 9:2847–53.
- Pennica D, Swanson TA, Shaw KJ, Kuang WJ, Gray CL, Beatty BG, et al. Human cardiotrophin-1: protein and gene structure, biological and binding activities, and chromosomal localization. Cytokine. 1996; 8:183–9.
- Stöckli KA, Lottspeich F, Sendtner M, Masiakowski P, Carroll P, Götz R, et al. Molecular cloning, expression and regional distribution of rat ciliary neurotrophic factor. Nature. 342:920–3.
- Liu J, Modrell B, Aruffo A, Scharnowske S, Shoyab M. Interactions between oncostatin M and the IL-6 signal transducer, gp130. Cytokine 1994, 6:272–8.
- Gearing DP, Bruce AG. Oncostatin M binds the high-affinity leukemia inhibitory factor receptor. New Biol. 1992; 4:61–5.
- Gearing DP, Comeau MR, Friend DJ, Gimpel SD, Thut CJ, McGourty J, et al. The IL-6 signal transducer, gp130: an oncostatin M receptor and affinity converter for the LIF receptor. Science. 1992; 255:1434–7.
- Mosley B, De Imus C, Friend D, Boiani N, Thoma B, Park LS, et al. Dual oncostatin M (OSM) receptors. Cloning and characterization of an alternative signaling subunit conferring OSM-specific receptor activation. J Biol Chem. 1996; 271:32635–43.
- Wollert KC, Taga T, Saito M, Narazaki M, Kishimoto T, Glembotski CC, et al. Cardiotrophin-1 activates a distinct form of cardiac muscle cell hypertrophy. Assembly of sarcomeric units in series VIA gp130/leukemia inhibitory factor receptor-dependent pathways. J Biol Chem. 1996; 271:9535–45.
- Heinrich PC, Behrmann I, Haan S, Hermanns HM, Müller-Newen G, Schaper F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J. 2003; 374:1–20.
- Rousseau F, Chevalier S, Guillet C, Ravon E, Diveu C, Froger J, et al. Ciliary neurotrophic factor, cardiotrophin-like cytokine, and neuropoietin share a conserved binding site on the ciliary neurotrophic factor receptor alpha chain. J Biol Chem. 2008; 283:30341–50.
- Port MD, Laszlo GS, Nathanson NM. Transregulation of leukemia inhibitory factor receptor expression and function by growth factors in neuroblastoma cells. J Neurochem. 2008; 106:1941–1951.
- Elson GC, Lelièvre E, Guillet C, Chevalier S, Plun-Favreau H, Froger J, et al. CLF associates with CLC to form a functional heteromeric ligand for the CNTF receptor complex. Nat Neurosci. 2000; 3:867–72.
- Stahl N, Yancopoulos GD. The tripartite CNTF receptor complex: activation and signaling involves components shared with other cytokines. J Neurobiol. 1994; 25:1454–66.
- Bartlett PF, Brooker GJ, Faux CH, Dutton R, Murphy M, Turnley A, et al. Regulation of neural stem cell differentiation in the forebrain. Immunol Cell Biol. 1998; 76:414-8.
- Gregg C, Weiss S. CNTF/LIF/gp130 receptor complex signaling maintains a VZ precursor differentiation gradient in the developing ventral forebrain. Development. 2005; 132:565-78.
- Kwon YW, Gurney ME. Systemic injections of ciliary neurotrophic factor induce sprouting by adult motor neurons. Neuroreport. 1994; 5:789-92.
- Wright MC, Son YJ. Ciliary neurotrophic factor is not required for terminal sprouting and compensatory reinnervation of neuromuscular synapses: re-evaluation of CNTF null mice. Exp Neurol. 2007; 205:437-48.
- Kurek JB, Bennett TM, Bower JJ, Muldoon CM, Austin L. Leukaemia inhibitory factor (LIF) production in a mouse model of spinal trauma. Neurosci Lett. 1998; 249:1-4.
- Derouet D, Rousseau F, Alfonsi F, Froger J, Hermann J, Barbier F, et al. Neuropoietin, a new IL-6-related cytokine signaling through the ciliary neurotrophic factor receptor. Proc Natl Acad Sci U S A. 2004; 101:4827-32.
- Lindhard A, Bentin-Ley U, Ravn V, Islin H, Hviid T, Rex S, et al. Biochemical evaluation of endometrial function at the time of implantation. Fertil Steril. 2002; 78:221–233.
- Allan EH, Hilton DJ, Brown MA, Evely RS, Yumita S, Metcalf D, et al. Osteoblasts display receptors for and responses to leukemia-inhibitory factor. J Cell Physiol. 1990; 145:110–9.
- Jay PR, Centrella M, Lorenzo J, Bruce AG, Horowitz MC. Oncostatin-M: a new bone active cytokine that activates osteoblasts and inhibits bone resorption. Endocrinology. 1996; 137:1151–8.
- Gough NM, Williams RL. The pleiotropic actions of leukemia inhibitory factor. Cancer Cells. 1989; 1:77–80.
- Majumder A, Banerjee S, Harrill JA, Machacek DW, Mohamad O, Bacanamwo M, et al. Neurotrophic effects of leukemia inhibitory factor on neural cells derived from human embryonic stem cells. Stem Cells. 2012; 30:2387–99.
- Nawa H, Nakanishi S, Patterson PH. Recombinant cholinergic differentiation factor (leukemia inhibitory factor) regulates sympathetic neuron phenotype by alterations in the size and amounts of neuropeptide mRNAs. J Neurochem. 1991; 56:2147–50.
- Kanazawa H, Ieda M, Kimura K, Arai T, Kawaguchi-Manabe H, Matsuhashi T, et al. Heart failure causes cholinergic transdifferentiation of cardiac sympathetic nerves via gp130-signaling cytokines in rodents. J Clin Invest. 2010; 120:408–421.
- Pennica D, King KL, Shaw KJ, Luis E, Rullamas J, Luoh SM, et al. Expression cloning of cardiotrophin 1, a cytokine that induces cardiac myocyte hypertrophy. Proc Natl Acad Sci U S A. 1995; 92:1142–1146.
- Turnley AM, Bartlett PF. Cytokines that signal through the leukemia inhibitory factor receptor-beta complex in the nervous system. J Neurochem. 2000; 74:889–899.
- Knight DA, D’Aprile AC, Spalding LJ, Goldie RG, Thompson PJ. Leukaemia inhibitory factor (LIF) upregulates excitatory non-adrenergic non-cholinergic and maintains cholinergic neural function in tracheal explants. Br J Pharmacol. 2000; 130:975–979.
- Rose TM, Bruce AG. Oncostatin M is a member of a cytokine family that includes leukemia-inhibitory factor, granulocyte colony-stimulating factor, and interleukin 6. Proc Natl Acad Sci U S A. 1991; 88:8641–8645.
- Rose TM, Lagrou MJ, Fransson I, Werelius B, Delattre O, Thomas G, et al. The genes for oncostatin M (OSM) and leukemia inhibitory factor (LIF) are tightly linked on human chromosome 22. Genomics. 1993; 17:136–140.
- Martinou JC, Martinou I, Kato AC. Cholinergic differentiation factor (CDF/LIF) promotes survival of isolated rat embryonic motoneurons in vitro. Neuron. 1992; 8:737–44.
- Oberle S, Schober A, Meyer V, Holtmann B, Henderson C, Sendtner M, et al. Loss of leukemia inhibitory factor receptor beta or cardiotrophin-1 causes similar deficits in preganglionic sympathetic neurons and adrenal medulla. J Neurosci. 2006; 26:1823–32.
- Gritman K, Winkle DM, Lorentz CU, Pennica D, Habecker BA. The lack of cardiotrophin-1 alters expression of interleukin-6 and leukemia inhibitory factor mRNA but does not impair cardiac injury response. Cytokine. 2006; 36:9–16.
- Pennica D, Swanson TA, Shaw KJ, Kuang WJ, Gray CL, Beatty BG, et al. Human cardiotrophin-1: protein and gene structure, biological and binding activities, and chromosomal localization. Cytokine. 1996; 8:183–189.
- Zhou D, Zheng X, Wang L, Stelmack G, Halayko AJ, Dorscheid D, et al. Expression and effects of cardiotrophin-1 (CT-1) in human airway smooth muscle cells. Br J Pharmacol. 2003; 140:1237–44.
- Zheng X, Zhou D, Seow CY, Bai TR. Cardiotrophin-1 alters airway smooth muscle structure and mechanical properties in airway explants. Am J Physiol Lung Cell Mol Physiol. 2004; 287:L1165–71.
- Dagoneau N, Bellais S, Blanchet P. Mutations in Cytokine Receptor-Like Factor 1 ( CRLF1) Account for Both Crisponi and Cold-Induced Sweating Syndromes. Am J Hum Genet. 2007; 80:966–970.
- Crisponi L, Crisponi G, Meloni A. Crisponi Syndrome Is Caused by Mutations in the CRLF1 Gene and Is Allelic to Cold-Induced Sweating Syndrome Type 1. Am J Hum Genet. 2007; 80:971–981.
- Ohno M, Kohyama J, Namihira M, Sanosaka T, Takahashi JA, Hashimoto N, et al. Neuropoietin induces neuroepithelial cells to differentiate into astrocytes via activation of STAT3. Cytokine. 2006; 36:17-22.
- Rigante D. Are there febrile diseases with a risk of sudden death in children? Arch Dis Child. 2012; 97:180.
- Herholz J, Meloni A, Marongiu M, Chiappe F, Deiana M, Herrero CR, et al. Differential secretion of the mutated protein is a major component affecting phenotypic severity in CRLF1-associated disorders. Eur J Hum Genet. 2011; 19:525–533.
- Sims NA. gp130 Signaling in bone cell biology: multiple roles revealed by analysis of genetically altered mice. Mol Cell Endocrinol. 2009; 310:30–9.
- Stanke M, Duong CV, Pape M, Geissen M, Burbach G, Deller T, et al. Target-dependent specification of the neurotransmitter phenotype: cholinergic differentiation of sympathetic neurons is mediated in vivo by gp 130 signaling. Development. 2006; 133:141–150.
- Alexander WS, Rakar S, Robb L, Farley A, Willson TA, Zhang JG, et al. Suckling defect in mice lacking the soluble haemopoietin receptor NR6. Curr Biol. 1999; 9:605–608.
- Forger NG, Prevette D, deLapeyrière O, Bovis B, Wang S, Bartlett P, et al. Cardiotrophin-like cytokine/cytokine-like factor 1 is an essential trophic factor for lumbar and facial motoneurons in vivo. J Neurosci. 2003; 23:8854–8858.
- Zhang F, Wen Y, Guo X. CRISPR/Cas9 for genome editing: progress, implications and challenges. Hum Mol Genet. 2014; 23(R1):R40-6.
- Veltrop M, Aartsma-Rus A. Antisense-mediated exon skipping: taking advantage of a trick from Mother Nature to treat rare genetic diseases. Exp Cell Res. 2014; 325:50-5.
- Karijolich J, Yu YT. Therapeutic suppression of premature termination codons: mechanisms and clinical considerations. Int J Mol Med. 2014; 34:355-62.
- Zingman LV, Park S, Olson TM, Alekseev AE, Terzic A. Aminoglycoside-induced translational read-through in disease: overcoming nonsense mutations by pharmacogenetic therapy. Clin Pharmacol Ther. 2007; 81:99-103.
- Finkel RS, Flanigan KM, Wong B, Bönnemann C, Sampson J, Sweeney HL, et al. Phase 2a study of ataluren-mediated dystrophin production in patients with nonsense mutation Duchenne muscular dystrophy. PLoS One. 2013; 8:e81302.
- Pibiri I, Lentini L, Melfi R, Gallucci G, Pace A, Spinello A, et al. Enhancement of premature stop codon readthrough in the CFTR gene by Ataluren (PTC124) derivatives. Eur J Med Chem. 2015; 101:236-44.
- Romeo Bertola D, Honjo RS, Baratela WA. Stüve-Wiedemann Syndrome: Update on Clinical and Genetic Aspects. Mol Syndromol. 2016; 7:12-8.
- Fokkema IF, Taschner PE, Schaafsma GC, Celli J, Laros JF, den Dunnen JT. Leiden Open Variation Database (LOVD) v.2.0: the next generation in gene variant databases. Hum Mutat. 2011; 32:557-63.