
The Female mdx Mouse: An Unexpected Vascular Story
Lorna Guéniot1a, Claire Latroche1ab, Cédric Thépenier1,2a, Laurent Chatre3,4, Aurélien Mazeraud1, Daniel Fiole1,2, Pierre L. Goossens1, Fabrice Chrétien1,5,6c and Gregory Jouvion1,5c
Abstract
Duchenne muscular dystrophy (DMD) is a progressive neuromuscular disease, caused by absence of functional dystrophin and inevitably leading to death. A variable proportion of women carriers (2.5 to 19%) can also manifest symptoms ranging from myalgia to cardiomyopathy, and pathophysiological mechanisms are still not completely understood. Our study focused on 12 month-old female mdx mice, displaying marked chronic muscle lesions, similar to the lesions observed in human DMD. Our aim was to focus on the alterations of the vascular network organisation, and functional repercussions using a combination of histology/morphometry techniques and totally non-invasive functional approach (multiparametric and functional nuclear magnetic resonance), clearly relevant for clinical diagnosis and research, combining arterial spin labeling imaging of perfusion, and 31P-spectroscopy of phosphocreatine kinetics. Collectively, our results demonstrate that the vasculature, both in its steady state organisation and dynamic behaviour after an ischemia-reperfusion stress, is altered in the 12 month-old female mdx mouse: increased density of vascular sections in histology, modification of the post-ischemic hyperemia profile, increase in mitochondrial oxidative rephosphorylation capacity, in striking opposition to what was observed in age-matched male mdx mice. We believe the apparent discordance between vascular and muscular features in the female mdx mouse make it an interesting tool to decipher further dystrophinopathy pathophysiological mechanisms.
Introduction
Duchenne muscular dystrophy (DMD) is an X-linked muscle disease arising from the absence of functional dystrophin protein. In 1 every 5000 birthed boys, this mutation leads to a progressive skeletal muscle weakness and cardiomyopathy, causing premature death with an approximate median life expectancy of 25 years1,2. Reportedly, 2.5 to 19% of women carriers can also be affected by symptoms ranging from myalgia to cardiomyopathies. Rare chromosomal aberrations have also been described to cause the full phenotype in female carriers3,4.
Dystrophin protein has been shown to have both a mechanical role and to act as a scaffold for other intracellular proteins. Mechanically, dystrophin links extracellular matrix to intracellular actin. This linkage allows force transmission and protects sarcolemmal membrane during muscle contraction. In dystrophic muscle, contraction leads to fiber damage triggering inflammation; regeneration cycles progressively lead to fibrosis and adipocyte infiltration5.
More recently, non-mechanical roles of dystrophin have partially been unveiled, partly stemming from its participation to a large intracellular complex, the dystrophin glycoprotein complex (DGC). Anchoring this complex, proteins such as NO-synthase (nNOS), aquaporin-4 (AQP4) or acetylcholine receptors are involved in the symptomatology and progressively turning DMD into a systemic disease6.
Skeletal muscle lesions have logically been the main focus of studies on DMD. Dystrophin expression is however not restricted to skeletal and myocardial muscle cells and has also been shown in endothelial or neuronal cells7. Before these discoveries, an involvement of the vasculature in the muscular phenotype had long been suspected. This originated from histological descriptions of grouped myofiber necrosis evoking a possible local ischemia, yet further studies were rather conflicting regarding any significant static microvascular defect in DMD patients8. The vascular hypothesis nevertheless survived and is currently included in a « two-hit » model, where lack of NO impairs local vasodilatation during muscular exercise, leading to a functional ischemia aggravating mechanical lesions. Both static and functional aspects have found support in experimental models where increased capillary density by Flt-1 (fms-like tyrosine kinase 1) knock-out, or increased NO pathway activity, translate into functional and histological benefit9,10.
We and others have recently shown that age explained part of conflicting data regarding vascular function in experimental models. Two month-old mdx male mice indeed display similar capillary density and enhanced angiogenic response to local ischemia in comparison to wild-type controls11. However, at 6 months, mdx mice display a decreased capillary density and angiogenic response to ischemia12. We recently showed that, in contrast to controls, steady-state microvasculature in skeletal muscle decreases with age in male mdx mice13. Abnormalities were not restricted to the capillary bed; short term reperfusion was markedly enhanced in young (3 months) male mdx mice compared to controls, while it was impaired in elder male mdx mice (12 months). This was consistent with the loss and disorganisation of terminal arterioles in muscle. This shed light on the multiple time and space scales at which vasculature should be studied in order to fully grasp its involvement in muscular demise.
Most experimental data have been gathered on male animals. Yet, studying gender effect on these models can lead to a better understanding of pathways involved in maintenance of tissue function, as well as testing of pathophysiological hypotheses. Age has already been shown to induce more functional decay in muscular function in female mdx mice than in their male counterpart14. This is consistent with the greater level of muscular damage and fibrosis found in aging female mdx mice compared to males, in the Gastrocnemius muscle15 and myocardium16. Conversely, in wild-type mice, the vascular dilatation response to exercise is preserved in aging females, but blunted in old males, the latter also re-constricting their vessels more rapidly after muscular exercise17.
How could these parameters, translated in mdx females, shed light on the reality of the vascular hypothesis as a direct cause of muscular wasting? Rare studies addressed the importance of the vascular network in the pathophysiology of dystrophinopathy in female. To tackle this question, we focused on the muscle vascular network in 12 month-old female mdx mice, both at morphological and functional levels using histological approaches compared to in vivo dynamic and non-invasive multiparametric and functional nuclear magnetic resonance (NMR) study.
Material and Methods
Mice
Female C57Bl/6J control mice were obtained from Charles River Laboratory (l’Arbresle, France), and female mdx-4Cv with C57Bl/6 background mice, model for human DMD, were kindly provided by Pr. Gherardi (Hôpital Henri Mondor, France). Twelve month-old animals were used. Animals were housed in animal facilities of the Institut Pasteur licensed by the French Ministry of Agriculture and complying with European Union regulations. Protocols were approved by the Institut Pasteur Animal Experimentation Ethics Committee (01332.02).
Nuclear Magnetic Resonance analysis
NMR experiments were performed on 12 monthold female mdx-4Cv (n = 7) and control C57Bl/6J (n = 6) mice. The experimental device was already described13. To highlight differences between normal and altered muscles, we applied a stress to increase the global need for perfusion. Ischemia-reperfusion stress was applied for 30 minutes to the mouse left hindlimb which provokes maximal vasodilatation and limited resistance of arteries/arterioles18 just after tourniquet release. During experiments, a water heating pad ensured a constant temperature of 37°C and breathing was monitored. Leg ischemia was induced by occlusion of femoral artery by two surgical threads placed around the thigh and pulled tight by application of a weight19. Efficiency of the occlusion protocol was assessed by Arterial Spin Labeling (ASL)- NMR imaging19. After a 30 min ischemia, the weight was removed, inducing a hyperaemic response monitored over the next 30 min. During the whole protocol, dynamic acquisitions of interleaved NMR scans of perfusion signal (ASL; time resolution: 10 s) and mitochondrial activity (31P-spectroscopy-NMRS; time resolution: 2.5 s) were collected using the dedicated Bruker MultiScanControl software (BrukerBioSpin GmbH)19,20.
In brief, ASL imaging is based on non-invasive alternate magnetic tagging of blood water spins to provide endogenous markers of muscle perfusion, measured in regions of interest (ROI) drawn in posterior compartment of the leg. Peak intensities and onset times have been calculated by a mean of each individual maximal reperfusion value and its associated onset time. Resting perfusion was calculated as the average of 260 s pre-occlusive values.
Muscle bioenergetics and pH were assessed from ratios of energetic phosphates measurable by 31P-NMRS at rest. In vivo mitochondrial oxidative capacity was directly assessed from the rate of creatine rephosphorylation at the end of ischemia. Intramuscular pH was calculated from chemical shift between phosphocreatine (PCr) and inorganic phosphate (Pi). A minimum of 50% PCr depletion at the end of ischemia was necessary to reliably measure dynamics for PCr recovery, and examinations which did not reach this threshold were rejected.
Histological/Immunofluorescence analysis
Gastrocnemius muscles were collected from mice after NMR experiments, snap frozen in liquid nitrogen-cooled isopentane and kept at –80°C. Four different levels of 7 μmthick sections were cut and stained with hematoxylin-eosin (HE) to describe histopathological modifications of muscle tissue, and Sirius red for visualisation of collagen. For immunofluorescence analyses, muscle cryosections were incubated with antibodies directed against endothelial cells (anti-CD31; BD Pharmingen), pericytes (anti-Neuron- Glial antigen 2 (NG2); Millipore), smooth muscle cells (αSMA; Sigma) and basal lamina (anti-laminin; Sigma). Briefly, primary antibodies were incubated overnight at 4°C and revealed by cy3- (αSMA) or TRITC-labeled (CD31, NG2, laminin) secondary antibodies (Thermo Scientific).
Morphometric analysis
Morphometric analysis was performed using ImageJ (NIH, Bethesda, MD, USA) and ZEN Lite 2012 (Zeiss, Jena, Germany) softwares. We evaluated: the percentage of collagen tissue in each muscle section (after Sirius red staining), distribution of muscle fiber diameter, percentage of centro- or peri-nucleated fibers, microvessel count and distribution around each myofiber and on the entire muscle section. Between 200 and 300 fibers were considered for each muscle.
MitoTracker and laminin co-immunostaining
Cryosections were incubated with MitoTracker Deep Red (Life technologies) for 1 hour then fixed with 2% paraformaldehyde (PFA; EMS) in cold PBS, permeabilized with 0.5% Triton X-100 for 10 min at 4 °C, washed and blocked with 5% BSA in PBS for 1 hour at room temperature. Sections were incubated with primary antibody rabbit anti-Laminin (Sigma-Aldrich) overnight at 4°C and with Alexa-conjugated secondary antibody and Hoechst 33342 for 1 hour at room temperature.
Three-dimensional confocal imaging and quantification of Mitotracker and Laminin fluorescence
Confocal acquisitions were performed using a spinning-disk Perkin-Elmer Ultraview RS Nipkow Disk, an inverted laser-scanning confocal microscope Zeiss Axiovert 200M with an Apochromat × 40/1.4 oil objective and a Hamamatsu ORCA II ER camera (Imagopole, PFID, Institut Pasteur). Optical slices were taken at 200-nm intervals along the z axis covering the whole depth of the cell, at a 1.024/1.024 pixel resolution. Three-dimensional reconstruction was achieved using the IMARIS software (Bitplane). Fluorescence quantification was done using a single-imaging frame. An average of 100 randomly chosen fibers were used for Mitotracker and Laminin quantification.
RT–qPCR
Total RNA was isolated from cells using the RNAeasy Mini kit (Qiagen), and reverse transcribed using Superscript IV Reverse transcriptase (Life Technologies). RT–qPCR was performed using Power Sybr Green PCR Master Mix (Applied Biosystems) and the rate of dye incorporation was monitored using the StepOne Plus RealTime PCR system (Applied Biosystems). Six and seven biological replicates were used for control wild-type (wt) and mdx mice, respectively. Data were analysed by StepOne Plus RT PCR software v2.1 and Microsoft excel. TBP transcript levels were used for normalization of each target (=ΔCT) (Table 1). RT–qPCR CT values were analyzed using the 2-(ΔΔCT) method to calculate the fold expression.
| Gene | Forward sequence | Reverse sequence |
|---|---|---|
| TBP | 5’-CTTCACCAATGACTCCTATG-3’ | 5’-TGACTGCAGCAAATCGCTTG-3’ |
| PGC-1α | 5’-GGACAGTCTCCCCGTGAT-3’ | 5’-TCCATCTGTCAGTGCATC-3’ |
| 16S rRNA | 5’-GGGACTAGCATGAACGGCTA-3’ | 5’-CCCCAACCGAAATTTCAAAC-3’ |
| CYTB | 5’-ACGTCCTTCCATGAGGACAA-3’ | 5’-GAGGTGAACGATTGCTAGGG-3’ |
Table 1: Oligonucleotide primers used for RT-qPCR.TBP: TATA box binding protein (nuclear reference); PGC-1α:PPARγ coactivator 1α; CYTB: cytochrome b.
Quantification of mtDNA content by qPCR
Total DNA was prepared using extraction buffer (0.2 mg ml−1 proteinase K, 0.2% SDS and 5 mM EDTA in PBS) and incubated at 50 °C for 3 h. DNA was precipitated with 3 M sodium acetate (pH 5.2) and isopropanol for 20 min on ice before centrifugation at 8,000g at 4 °C. The DNA pellet was washed and briefly air dried. qPCR amplification was performed on total DNA using the StepOne Plus RealTime PCR system (Applied Biosystems) and Power Sybr Green PCR Master mix (ABI) following the manufacturer’s instruction. A fragment of mitochondrial CO1 gene, the established marker for mtDNA content in mouse cells, was amplified using the nuclear encoded Ndufv1 gene as endogenous reference (Table 2). The level of mtDNA was calculated using the ΔCT of average CT of mtDNA and nDNA (ΔCT=CT nDNA−CT mtDNA) as 2-(ΔCT).
>
| Gene | Forward sequence | Reverse sequence |
|---|---|---|
| NDUFV1 | 5’- CTTCCCCACTGGCCTCAAG-3’ | 5’- CCAAAACCCAGTGATCCAGC-3’ |
| CO1 | 5’- TGCTAGCCGCAGGCATTAC-3’ | 5’- TGCTAGCCGCAGGCATTAC-3’ |
Table 2: Oligonucleotide primers used for qPCR.NDUFV1: NADH dehydrogenase (ubiquinone) flavoprotein 1 (nuclear reference); CO1: mitochondrially encoded cytochrome c oxidase 1 (mitochondrial DNA).
Statistics
Group comparisons for perfusion parameters and phosphorus spectroscopy analysis were performed using Mann-Whitney U test. Coefficients from linear regressions and linear regressions taking into account qualitative variables (Covariance Analysis, ANCOVA) were tested against 0 by resampling, using lmPerm package of R (version 3.2.2; https://cran.r-project.org/) running on RStudio (version 0.99.473; https://www.rstudio.com/).
Statistical analysis of histological data was performed with GraphPad-Prism software (La Jolla, CA, USA). Fiber diameter repartition was evaluated by a chi-square test followed by a multi-t-test corrected for multiple comparisons using Holm-Sidak method. Same multi-t-test was used to evaluate capillary count/fiber repartition.
Statistical significance was taken at p < 0.05 and p-values indicated on figures are *p < 0.05, **p < 0.01, and ***p < 0.001. Numerical NMR, histological, RT-qPCR and qPCR data are reported as mean ± SD. Quantification of fluorescence activity is reported as mean ± SEM.
Results
Characterization of the Gastrocnemius muscle tissue, as expected in 12 month-old female mdx mice, showed a polyphasic multifocal lesion, characterized by mild endomysial/perimysial fibrosis, small inflammatory infiltrates (macrophages mostly), anisocytosis, and centrally nucleated fibers (Figure 1). Muscles in control 12 month-old wild-type C57BL/6 mice did not display any lesion.
Post-ischemic muscle blood perfusion is altered in female mdx mice.
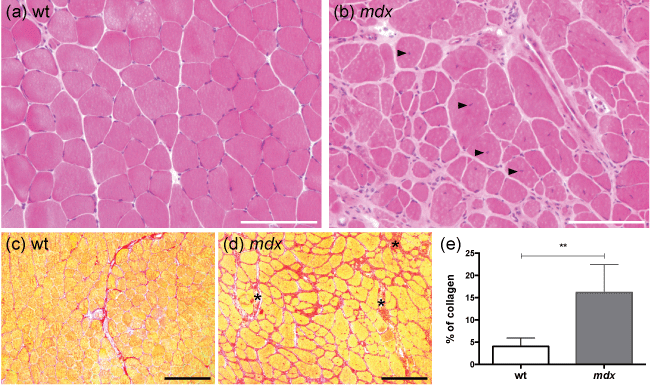
Figure 1: Histopathological characterisation of Gastrocnemius muscle in 12-month-old female mdx mice.
mdx mice displayed chronic histological lesions, characterized by multifocal inflammatory infiltrates (mostly macrophages), marked anisocytosis with the presence of atrophic and regenerating myofibers (displaying centrally located nuclei; b-black arrowheads), and endomysial/perimysial fibrosis (d-black stars, e). Wild-type mice displayed histologically normal muscles (a) with no fibrosis (c, e). a, b: hematoxylin and eosin staining; c, d: Sirius red staining (specific for collagen); ** p < 0.01; scale bar = 100 μm (a, b), scale bar = 200 μm (c, d); n = 6 (wt, e), n = 7 (mdx, e).
Gastrocnemius muscle perfusion was explored through an in vivo dynamic and non-invasive multiparametric and functional NMR study. No difference was observed at rest between female mdx and wild-type mice (wt: 5.3 ± 2.3 mL/ min/100 g; mdx: 6.2 ± 0.2 mL/min/100 g).
Blood vessel function was then challenged through an ischemia-reperfusion protocol to highlight perfusion differences. After 30 minutes of ischemia, the release of tourniquet provoked an instantaneous increase of perfusion in both groups (Figure 2a). However, this increase was significantly higher in mdx female mice for the first 25 minutes (Table 3).
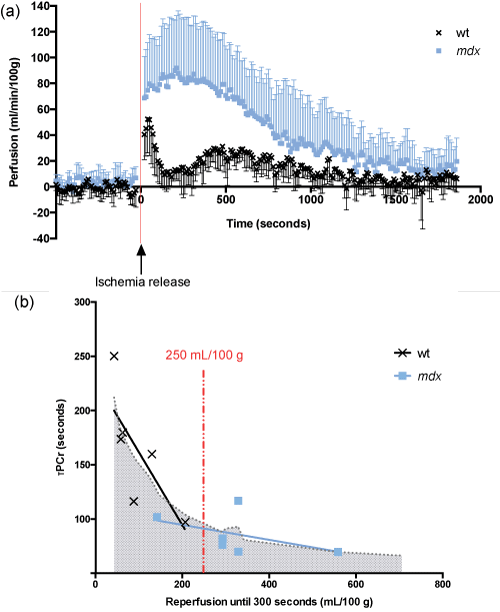
Figure 2: Alteration of reperfusion profiles after 30 minutes of ischemia in 12-month-old female mdx mice.
| Variable | Wild-type mice | mdx mice |
|---|---|---|
| Basal perfusion (mL/min/100 g) | 5.3 ± 2.3 | 6.2 ± 0.2 |
| Reperfusion mean (mL/min/100g) | 16.2 ± 4.8 | 49.7 ± 29.2** |
| First peak | ||
| Onset time (s) | 38 ± 12 | 166 ± 93** |
| Intensity (mL/min/100 g) | 55.9 ± 24.6 | 102.3 ± 41.3 |
| Time to half-constriction (s) | 72 ± 16 | 463 ± 23** |
| Second peak | ||
| Onset time (s) | 450 ± 35 | None |
| Intensity (mL/min/100 g) | 41.4 ± 5.9 | None |
Table 3: Perfusion analysis from 1H-NMR in 12-month-old female mdx mice.
Reperfusion mean has been calculated during the first 25 minutes after release of ischemia. Wild-type mice, n=6; mdx mice, n=7. **p < 0.01.
(a)The set-up of the tourniquet was sufficient to induce an absence of perfusion in both groups. After release of tourniquet, a rapid and important increase of perfusion was detected and the reperfusion of mdx mice was greater than wild-type mice. A single peak of reperfusion was however observed in mdxmice, when a first rapid and strong peak followed by a second attenuated peak of reperfusion was observed in wild-type animals.
Because the release of ischemia induced movements of the leg, images affected by these movement artifacts, at the moment of ischemia release, were removed from analysis of muscle perfusion.(b) In the first 5 minutes after ischemic stress release, below a threshold of 250 mL/100 g reperfusion (concerning mostly wt mice), PCr resynthesis rate was dependent on perfusion, an increase in reperfusion leading to a decrease of τPCr. In contrast, above the threshold of 250 mL/100 g reperfusion (concerning mostly mdx mice), PCr resynthesis rate was poorly affected by the increase of post-ischemia reperfusion.
a: Perfusion analysis was performed through from 1H-NMR as described in the Material and Methods section; b: Correlation between reperfusion and PCr resynthesis rate (from 31P-spectroscopy analysis) during the first 300 seconds (5 minutes) after ischemia; n = 6 (wt, a, b); n = 7 (mdx, a, b); τPCr: time of creatine rephosphorylation.
Wild-type and mdx mice also displayed different profiles of re-perfusion, with specific differences in the early phase (Table 3). Wild-type mice indeed displayed an initial peak of perfusion at 38 s after the tourniquet release, followed by a second attenuated and larger peak of reperfusion at 450 s. In contrast, mdx mice reperfusion profile did not present a biphasic morphology, but a single delayed and larger peak of reperfusion at 166 s post-ischemia.
Muscle bioenergetics was consistently altered in female mdx mice.
Female mdx mice displayed marked alterations in muscle bioenergetics parameters (Table 4). At rest, mdx mice displayed a higher Pi/PCr ratio compared to wildtype mice, which reflects an increase in ADP concentration in dystrophic mice. The Pi/PCr ratio was still higher in mdx mice during the ischemic stress, which also induced an accelerated PCr depletion (ΔPCr) compared to wild-type mice.
| Experimental condition | Variable | Wild-type mice | mdx mice |
|---|---|---|---|
| Rest | pH | 7.15 ± 0.06 | 7.19 ± 0.04 |
| Pi/PCr | 0.07 ± 0.02 | 0.10 ± 0.01** | |
| PCr/ATPγ | 3.34 ± 0.31 | 3.08 ± 0.15 | |
| End of ischemia | pH | 6.95 ± 0.03 | 6.91 ± 0.06 |
| Pi/PCr | 0.93 ± 0.21 | 1.37 ± 0.31* | |
| PCr/ATPγ | 2.33 ± 1.67 | 1.25 ± 0.44 | |
| τPCr (s) | 163 ± 54 | 83 ± 20** |
Table 4: Energetic metabolism alterations in 12-month-old female mdx mice.Energetic metabolism analysis was performed through 31P-spectroscopy as described in the Material and Methods section. The mean depletion of phosphocreatine, ΔPCr (%), for wt and mdx mice (55 ± 2 and 62 ± 4**,respectively) was above 50%, confirming the efficiency of the ischemic stress. Wild-type mice, n=6; mdx mice, n=7.
* p< 0.05, ** p < 0.01.
PCr: phosphocreatine; Pi: inorganic phosphate; τPCr: time of creatine rephosphorylation.
During reactive hyperemia after tourniquet release, a
significant acceleration of PCr resynthesis rate (τPCr) was observed in mdx mice compared to wild-type mice, reflecting higher mitochondrial ATP production in mdx mice.
Phosphocreatine depletion was thus accelerated during ischemia in mdx female mice, and, at recovery, mitochondrial oxidative rephosphorylation was unexpectedly faster and perfusion was increased in comparison to control mice.
Combined analysis of our 31P-NMRS and perfusion data revealed a biphasic association between PCr resynthesis rate and reperfusion during the first 300 seconds (Figure 2b).
The linear regression slopes of τPCr on perfusion area under the curve (AUC) was significantly different above and below a reperfusion value of 250 mL/100 g (p < 0.05). Below this threshold, PCr resynthesis rate was indeed dependent on perfusion; each 1 mL/min/100 g increase in perfusion decreased the PCr resynthesis time of 3.55 seconds. Conversely, no further significant modification of the PCr resynthesis rate was observed above 250 mL/100 g of post-ischemia reperfusion.
Alongside this higher mitochondrial metabolism in both resting and post-ischemic states, female mdx mice displayed a significant increase in laminin and in mitochondrial mass measured by MitoTracker compared to wild-type female mice (Figure 3).
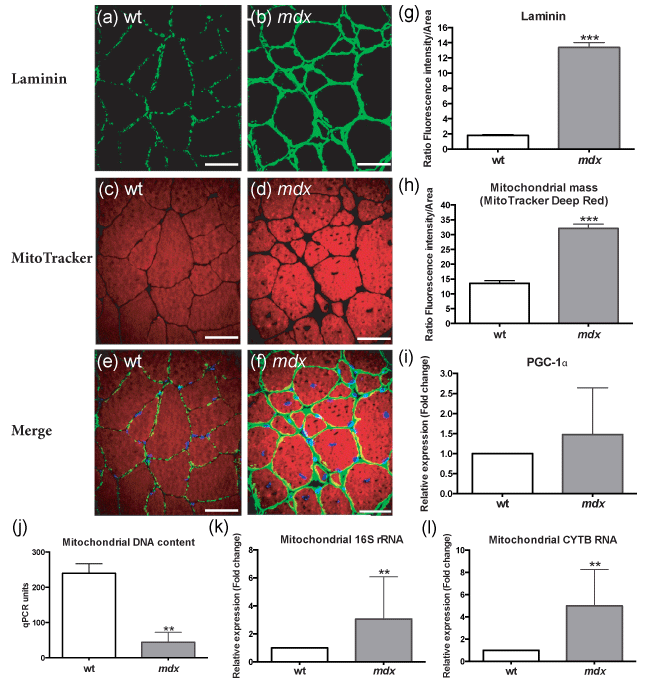
Figure 3: Mitochondrial function was altered in 12 month-old female mdx mice.
(a-h): mdx mice displayed an increased laminin and mitochondrial mass compared to wild-type mice.
(i): Despite mitochondrial mass difference, PGC-1α was similar between both groups.
(j-l): Muscles from mdx mice displayed a loss in mitochondrial DNA (mtDNA) not affecting mitochondrial RNA (mtRNA) expression which was increased comparatively to wild-type mice.
a-f: Confocal microscopy 3D-reconstructed laminin and mitochondria distribution in wild-type and mdx muscle fibers.
g-h: Quantification of fluorescence intensity normalized to fiber area. Quantification by i: RT-qPCR of PGC-1α, j: qPCR of mtDNA content and k-l: RT-qPCR of 16S and cytochrome b (CYTB) mitochondrial RNAs; * p < 0.05; ** p < 0.01; *** p < 0.001; scale bar = 50 μm (a-f); n = 6 (wt, a-l); n = 7 (mdx, a-l).
Surprisingly, the transcriptional coactivator PGC-1α, a master regulator of laminin, mitochondrial biogenesis, ROS homeostasis, oxidative metabolism and muscle function21, did not increase its expression. Mitochondrial activity strongly depends on mitochondrial DNA (mtDNA) and RNAs (mtRNAs). Muscles from female mdx mice lost most of their mtDNA which confirmed mitochondrial alteration. Intriguingly, the depletion of mtDNA was uncoupled to the levels of mtRNAs suggesting the maintenance of a huge mitochondrial transcriptional activity despite mtDNA depletion (Figure 3). This discordant phenotype between mitochondrial mass, DNA and transcriptional activity is suggestive of a muscular mitochondrial and metabolic alteration in 12 month-old female mdx mice.
Alteration of the vascular network organisation in female mdx mice.
The functional alterations observed in mdx mice prompted us to explore the microvascular network in the Gastrocnemius muscle.
Immunofluorescence analyses showed that: (i) myofiber cross-section mean diameter was smaller in mdx mice, (ii) the smallest myofibers (diameter < 40 μm) represented more than 50% of total muscle fibers in mdx mice in contrast to 30% in controls and (iii) these small myofibers were undervascularized (Figure 4).
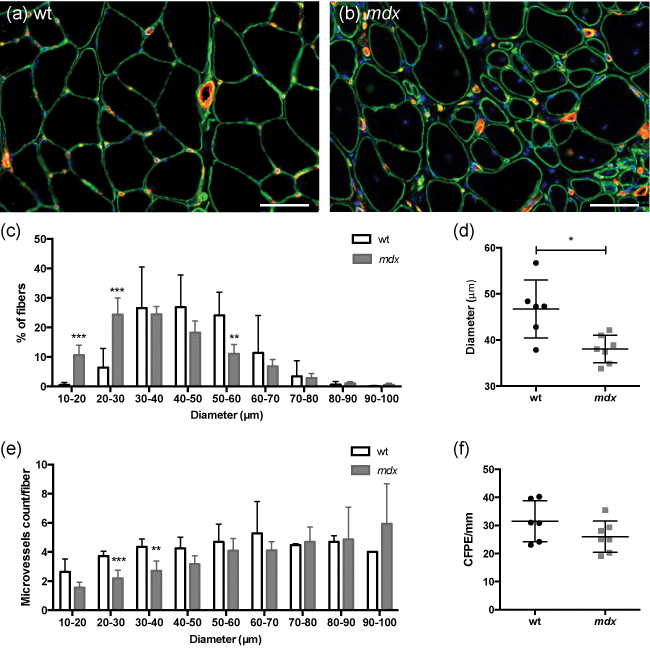
Figure 4: 12 month-old female mdx mice displayed myofiber size heterogeneity with a modified distribution of microvessels.
Anisocytosis was more severe in mdx mice and characterized by a smaller mean fiber size (a-d). Atrophic myofibers with a diameter under 40 μm represented more than 50 % of the total myofibers in mdx mice (c). These atrophic myofibers displayed fewer microvessels at their periphery (b, e). The distance between microvessels and myofibers, calculated using the capillary-to-fiber perimeter exchange index (CFPE) was similar in both groups (f).
a, b: laminin-fluorescein isothiocyanate (FITC) and CD31-indocarbocyanine (Cy3) immunofluorescence labeling of basal lamina (green) and blood vessels (red); * p < 0.05; ** p < 0.01; *** p < 0.001; scale bar = 50 μm (a-b); n = 6 (wt, a-f); n = 7 (mdx, a-f).
To further characterize the microvascular network, we evaluated the density (per mm2) of microvessels (defined as CD31+ vascular structures, with a diameter < 20 μm) and terminal arterioles (defined as NG2+/αSMA+ perivascular cells, with a diameter < 20 μm) in female mdx mice. We observed an increase in microvessel density, but no difference in terminal arteriole density (Figure 5a-e). This is in contrast to what has been reported in age-matched male mdx mice13. However, a decrease in pericyte density (NG2+ cells located at the periphery of blood vessels in muscle sections)23 was detected in female mdx mice (wt: 399 ± 65 pericytes/mm2; mdx: 298 ± 64 pericytes/mm2; p < 0.01) (Figure 5c).
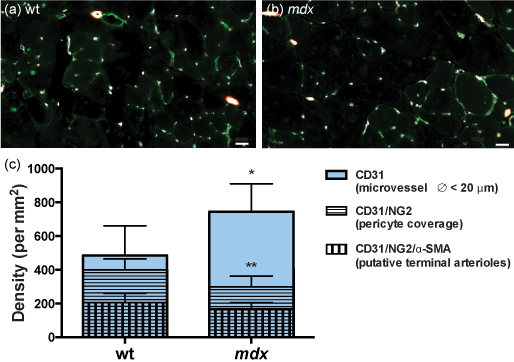
Figure 5: More microvessels but a modification of microvascular coverage in 12 month-old female mdx mice.
In comparison to wild-type mice, mdx mice displayed more microvessels per mm2, fewer pericytes, and a similar density of small terminal arterioles (Ø < 20 μm) (a-c).
a, b: CD31-indodicarbocyanine (Cy5), NG2-fluorescein isothiocyanate (FITC) and α-SMA-indocarbocyanine(Cy3) immunofluorescence to label blood vessels (white), pericytes (green) and smooth muscle cells (red); * p < 0.05; ** p < 0.01; scale bar = 20 μm (a, b); n = 6 (wt,c); n = 7 (mdx, c).
At the macrovascular scale - i.e. for blood vessels with a section diameter > 20 μm, we detected an increase in arteriole density (CD31+/α-SMA+) in female mdx mice in comparison to wild-type mice (wt: 6.0 ± 0.4 arterioles/ mm2; mdx: 10.4 ± 2.0 arterioles/mm2; p < 0.01) (Figure 6).
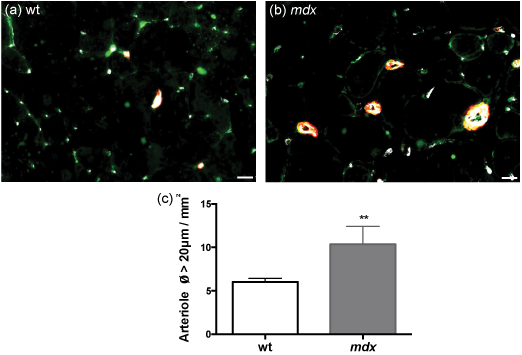
Figure 6: Increased muscular arteriole (Ø > 20 μm) density in 12 month-old female mdx mice.
mdx mice displayed more arterioles (Ø > 20 μm) than wild-type mice (a-c).
a, b: CD31-indodicarbocyanine (Cy5), NG2-fluorescein isothiocyanate (FITC) and α-SMA-indocarbocyanine (Cy3) immunofluorescence to label blood vessels (white), pericytes (green) and smooth muscle cells (red); ** p < 0.01; scale bar = 20 μm (a, b); n = 6 (wt, c); n = 7 (mdx, c).
Discussion
In this study, we investigated the in vivo impact of mdx phenotype on skeletal muscle vasculature and metabolism in the 12 month-old female mdx mouse, extending our previous results obtained in male mdx mice13. Collectively, our results demonstrate that the vasculature, both in its steady state organization and dynamic behavior after an ischemia-reperfusion stress, is altered in femalemdx mouse: increased density of vascular sections in histology, modification of the post-ischemic hyperemia profile, increase in overall mitochondrial oxidative rephosphorylation capacity, in striking opposition to what was observed in age-matched male mdx mice.
Histologically, we found a more severe muscle fibrosis in 1 year-old female mdx mice in comparison to wild-type mice with about 16% of collagen deposition, whereas agematched male mice showed a 10% in our previous study in aging male mdx mice13. These data fit the literature describing the evolution of muscle fibrosis in mdx mice, increasing over time16, yet differentially affecting both genders. Salimena et al. indeed showed a trend towards less fibrosis in young (3 month-old) mdx females than in mdx males, and conversely a more severe fibrosis in 1 yearold females than in males14,15.
The level of collagen deposition we found in female mdx mice remained moderate, with endomysial fibrosis having no repercussion on the distance between capillaries and myofibers (no modification of the CFPE index), thereby allowing the study of perfusion by arterial spin labeling without inducing a significant bias.
Reperfusion blood flow revealed two main differences between mdx and wt females. (i) The maximal blood flow after tourniquet release was significantly increased in mdx females compared to wt. This measure, indicative of maximal or sub-maximal vasodilatation19, could be considered an emerging feature from several passive parameters including vascular maximal diameter (which also depends on the maximal number of recruited blood vessels), vascular elasticity as well as perivascular stiffness24. (ii) The return to basal perfusion after this first maximum was also evidently increased in mdx females suggesting a possible impaired function of active components of vasoconstriction (pericytes, smooth muscle cells, vascular innervation).
Interestingly, the implications of these perfusion results, i.e. increased maximal vasodilatation and decreased vasomotor tone, are consistent with data gathered by Mancinelli et al. Their study described the ex vivo behavior of portal veins from 8 month-old mice when longitudinally stretched. Portal vein displays, as arteries and arterioles, a layer of smooth muscular cells. Contrasting with wt and male mdx mice, female mdx showed a lower increase in tension to passive stretch, and a marked disorganization of spontaneous contractile activity. The authors concluded that female mdx mice presented an increased distensibility of vessel wall, associated with irregular contractions, likely arising from functional segmentation of the vessel by altered intercellular communications25.
Our histological analysis of vascular actors yielded a possible anatomical explanation of perfusion dynamics. Female mdx mice presented an increase in vascular structures per surface unit (both arterioles with diameter > 20 μm and microvessels), associated with a decreased coverage by NG2 labeled cells around microvessels.
Literature about gender effect on vasculature in mouse models further indicate that oestrogens directly inhibit vascular smooth muscle cell proliferation26 and contraction27, as well as the pro-constriction vascular α1 adrenergic receptor expression28. This may account for the longer time to dilatation and increased speed of vasoconstriction described in muscle arterioles in 2 yearold males versus females during exercise17. As a whole, this literature points towards a decreased sympathetic tone associated with a decreased ability to respond to a given sympathetic stimuli in female mice, corroborating the vascular phenotype of premenopausal women29.
Concerning the impact of dystrophin absence on vasculature, several studies, usually performed using male subjects, demonstrated an impairment of perfusion, due to a dysfunction of vasodilatation. This dysfunction was consistently observed in the mdx mouse30-32, Golden Retriever Muscular Dystrophy dog33, and Human34, suggesting the existence of a “functional muscle ischemia” hypothesis in dystrophic muscles8. Interestingly, each actor implicated in vascular tone shows an abnormal behavior in the dystrophic context: (i) Endothelial cells from male mdx mice present an altered migratory behavior, an increased ex vivo apoptosis as well as a lesser ability to support the so-called angiogenic-myogenic coupling12, and (ii) vascular smooth muscle cells from dystrophic animals show less contractile activity to ex vivo stimulation by KCl35, possibly through a decreased functionality of calcic ryanodin receptors36. (iii) Dystrophin has also been shown to stabilize post-synaptic acetylcholine receptors. Its absence induces neuromuscular junction abnormalities, as well as decreased survival of sympathetic neurons as their ganglion communication (and thus their survival) relies on acetylcholine37. Nevertheless several studies, relying mostly on heart rate variability as an indirect marker, have concluded on an increase in sympathetic tone during DMD in human and mice38.
The phenotype described in our experiments sharply contrasts with the impaired vasodilatation found in the aforementioned literature focusing mostly on males.
Factors involved in this gender effect could involve (i) central or peripheral modulation of the sympathetic nervous systems, (ii) direct inhibition of vascular smooth muscle cell contractility, or (iii) enhancement of vasodilatory signalling. The decrease in microvascular coverage by αSMA positive cells, in our study, argues for a direct involvement of perivascular cells, either by a decrease in number or a lack of expression of contractile proteins including αSMA. Yet the observed decreased number of perivascular αSMA-expressing cells in 1 year-old male mdx mice does not parallel a decrease in vascular tone13, possibly implying that gender effect might target more the function than the number of vascular actors. Whether local loss of perivascular cells is due to detachment and participates in inducing a pro-fibrotic phenotype in these cells remains to be determined39.
The dynamic flow parameters described for female mdx (higher reperfusion peak and delay to return to basal flow in our experiments) logically translate into an increase in global reperfusion in comparison to wildtype females. Female mdx mice additionally show shorter τPCr, indicative of a higher maximal mitochondrial ATP synthesis. This result is indicative of a metabolism relying more heavily on its mitochondria for ATP synthesis in mdx female mice than in wild-type. Such a metabolic switch is consistent with the shift from fast glycolytic to the more oxidative fibers, a phenomenon already described in the permanently regenerating muscles of both mdx and utrophin/dystrophin KO mice40-42. This overall higher mitochondrial capacity in the Gastrocnemius muscle of mdx female mice does not preclude that these mitochondria be individually functionally impaired, as has already been described in dystrophinopathy models41. The discrepancy between an elevated level of MitoTracker labeling and a lower amount of mitochondrial DNA in mdx mice argues for large dysfunctional mitochondria, consistent with the swollen mitochondria described in Tibialis anterior from 3 month-old mdx mice43.
In our experiments, metabolism and perfusion parameters appear associated, τPCr showing a threshold association with post-ischemia reperfusion. Below a mean reperfusion of 250 mL / 100 g during the first 5 min posttourniquet release, an increase in perfusion is associated with the shortening of τPCr, whereas above that threshold (concerning most of the mdx mice), this association no longer exists; an increase in perfusion has therefore no impact on the τPCr anymore, probably illustrating the existence of a “luxury perfusion” above the threshold. Thus, the increase in post-ischemia reperfusion seen in mdx females might lead to a beneficial increase in mitochondrial bioenergetics during reperfusion, but, at least on our measured parameters, there is a maximum to this benefit.
Conclusion
In conclusion, dystrophin deficiency induces specific alterations of vascular structure and reactivity in 1 yearold female mice. Main features include an increase in blood vessel density (microvessels and arterioles), associated with an increase in reperfusion after ischemic stress, possibly overreaching local needs. This increase in vascular compliance and decrease in vascular tone are markedly different from the effect of dystrophin deficiency in male mdx mice. Our observations would be unexpected from a “functional ischaemia” hypothesis perspective, therefore raising the interesting question of their translation to a muscular exercise context.
Whether fibrosis partly results from functional ischemia, vascular abnormalities, or whether both could be common consequences of a third party actor (autonomous nervous system or perivascular cells) remains to be determined. We believe the apparent discordance between vascular and muscular features in the female mdx mouse make it an interesting tool to decipher further dystrophinopathy pathophysiological mechanisms.
List of abbreviations
ADP: Adenosine diphosphate, ASL: Arterial spin labeling, ATP: Adenosine triphosphate, CFPE: capillary to fiber perimeter exchange, CO1: mitochondrially encoded cytochrome c oxidase 1, CYTB: cytochrome b, DGC: Dystrophin glycoprotein complex, DMD: Duchenne muscular dystrophy, HE: haematoxylin and eosin, NDUFV1: NADH dehydrogenase (ubiquinone) flavoprotein 1, NG2: neuronglial antigen 2, NMR: nuclear magnetic resonance, NOS: nitric oxide synthase, PCr: Phosphocreatine, PGC-1α: PPARγ coactivator 1α, Pi: phosphate inorganic, SATIR: SATuration- Inversion Recovery, TBP: TATA box binding protein.
Authors’ contributions
GJ, FC, LG, and CL designed experiments. CL, LG, DF and LC carried out experiments (except NMR experiments) and GJ, FC, CT, CL, LG, DF and LC analysed data. CT, LG, GJ, PLG, CL, and LC were involved in writing the paper and all authors gave final approval of the submitted and published versions.
Acknowledgements
We would like to thank Anne Danckaert (Institut Pasteur) for the precious help in biostatistical analyses, Patricia Flamant and David Briand (Institut Pasteur) for their technical support for the histopathological experiments, Béatrice Matot, Aurea Martins-Bach, and Pierre G. Carlier (Institut de Myologie – CEA) for the NMR experiments, and Nathalie Denoyes (Institut Pasteur) for the support and encouragement. We thank the Imagopole (PFID) facility of Institut Pasteur for their help in mitochondrial imaging. This work was supported by a grant from the Association Française contre les Myopathies (AFM).
References
- Mendell JR, Shilling C, Leslie ND, Flanigan KM, al-Dahhak R, Gastier- Foster J, et al. Evidence-based path to newborn screening for duchenne muscular dystrophy. Ann Neurol. 2012; 71: 304–313.
- Passamano L, Taglia A, Palladino A, Viggiano E, D’Ambrosio P, Scutifero M, et al. Improvement of survival in Duchenne Muscular Dystrophy: retrospective analysis of 835 patients. Acta Myol. 2012; 31: 121–5.
- Lee SH, Lee JH, Lee KA, Choi YC. Clinical and genetic characterization of female dystrophinopathy. J Clin Neurol. 2015; 11: 248–251.
- Magot A, Mercier S, Péréon Y. Particularités de la dystrophie musculaire de Becker et des femmes conductrices. Arch Pédiatrie. 2015; 22: 12S31-12S36.
- De Paepe B, De Bleecker JL. Cytokines and chemokines as regulators of skeletal muscle inflammation: Presenting the case of Duchenne muscular dystrophy. Mediators Inflamm. 2013: 540370.
- Nichols B, Takeda S, Yokota T. Nonmechanical Roles of Dystrophin and Associated Proteins in Exercise. Neuromuscular Junctions. and Brains. Brain Sci. 2015; 5: 275–298.
- Di Angelantonio S, De Stefano ME, Piccioni A, Lombardi L, Gotti C, Paggi P. Neurobiology of disease lack of dystrophin functionally affects α3β2/β4-nicotinic acethylcholine receptors in sympathetic neurons of dystrophic mdx mice. Neurobiol Dis. 2011; 41: 528–537.
- Thomas GD. Functional muscle ischemia in Duchenne and Becker muscular dystrophy. Front Physiol. 2013; 4: 1–6.
- Thomas GD, Ye J, De Nardi C, Monopoli A, Ongini E, Victor RG. Treatment with a nitric oxide-donating NSAID alleviates functional muscle ischemia in the mouse model of Duchenne muscular dystrophy. PLoS ONE. 2012; 7: 3–10.
- Verma M, Asakura Y, Hirai H, Watanabe S, Tastad C, Fong GH. Flt-1 haploinsufficiency ameliorates muscular dystrophy phenotype by developmentally increased vasculature in mdx mice. Hum Mol Genet. 2010; 19: 4145–4159.
- Straino S, Germani A, Di Carlo A, Porcelli D, De Mori R, Mangoni A. Enhanced arteriogenesis and wound repair in dystrophin-deficient mdx mice. Circulation. 2004; 110: 3341–3348.
- Palladino M, Gatto I, Neri V, Straino S, Smith RC, Silver M, et al. Angiogenic impairment of the vascular endothelium: a novel mechanism and potential therapeutic target in muscular dystrophy. Arterioscler Thromb Vasc Biol. 33: 2867–2876.
- Latroche C, Matot B, Martins-Bach A, Briand D, Chazaud B, Wary C. Structural and functional alterations of skeletal muscle microvasculature in dystrophin-deficient mdx mice. Am J Pathol. 2015; 185: 2482–2494.
- Hourdé C, Joanne P, Noirez P, Agbulut O, Butler-Browne G, Ferry A. Protective effect of female gender-related factors on muscle forcegenerating capacity and fragility in the dystrophic mdx mouse. Muscle Nerve. 2013; 48: 68–75.
- Salimena MC, Lagrota-Candido J, Quírico-Santos T. Gender dimorphism influences extracellular matrix expression and regeneration of muscular tissue in mdx dystrophic mice. Histochem Cell Biol. 2004; 122: 435–444.
- Hakim CH, Duan D (2012). Gender differences in contractile and passive properties of mdx extensor digitorum longus muscle. Muscle Nerve. 2012; 45: 250–256.
- Bearden SE. Advancing age produces sex differences in vasomotor kinetics during and after skeletal muscle contraction. Am. J. Physiol. Regul Integr Comp Physiol. 2007; 293: R1274–R1279.
- Bertoldi D, Parzy E, Fromes Y, Wary C, Leroy-Willig A, Carlier PG. New Insight into Abnormal Muscle Vasodilatory Responses in Aged Hypertensive Rats by in vivo Nuclear Magnetic Resonance Imaging of Perfusion. J Vasc Res. 2006; 43: 149–156.
- Bertoldi D, Loureiro de Sousa P, Fromes Y, Wary C, Carlier PG. Quantitative, dynamic and noninvasive determination of skeletal muscle perfusion in mouse leg by NMR arterial spin-labeled imaging. Magn Reson Imaging. 2008; 26: 1259–1265.
- Baligand C1, Gilson H, Ménard JC, Schakman O, Wary C, Thissen JP et al. Functional assessment of skeletal muscle in intact mice lacking myostatin by concurrent NMR imaging and spectroscopy. Gene Ther. 2010; 17:328–337.
- Austin S, St-Pierre J. PGC1 and mitochondrial metabolism - emerging concepts and relevance in ageing and neurodegenerative disorders. J Cell Sci. 2012; 125: 4963–4971.
- Hepple RT. A new measurement of tissue capillarity: the capillary-tofibre perimeter exchange index. Can J Appl Physiol. 1997; 22: 11–22.
- Wanjare M, Kusuma S, Gerecht S. Perivascular cells in blood vessel regeneration. Biotechnol J. 2013; 8: 434–447.
- Wheeler JB, Mukherjee R, Stroud RE, Jones JA, Ikonomidis JS. Relation of murine thoracic aortic structural and cellular changes with aging to passive and active mechanical properties. J Am Heart Assoc. 2015; 4: 1–10.
- Mancinelli R, Tonali P, Romani R, Tringali A, Vargiu R, AZZENA GB. Mechanical properties of smooth muscle portal vein in normal and dystrophin-deficient (mdx) mice. Exp Physiol 1999; 84: 929–940.
- Sivritas D, Becher MU, Ebrahimian T, Arfa O, Rapp S, Bohner A, et al. Antiproliferative effect of estrogen in vascular smooth muscle cells is mediated by Kruppel-like factor-4 and manganese superoxide dismutase. Basic Res Cardiol. 2011; 106: 563–575.
- Ma Y, Qiao X, Falone AE, Reslan OM, Sheppard SJ, Khalil RA. Genderspecific reduction in contraction is associated with increased estrogen receptor expression in single vascular smooth muscle cells of female rat. Cell Physiol. Biochem. 2010; 26: 457–470.
- Zhang Y, Davidge ST. Effect of Estrogen Replacement on Vasoconstrictor Responses in Rat Mesenteric Arteries. Hypertension. 1999; 34: 1117– 1122.
- Joyner MJ, Wallin BG, Charkoudian N. Sex differences and blood pressure regulation in humans. Exp Physiol. 2016; 101: 349–355.
- Asai A, Sahani N, Kaneki M, Ouchi Y, Martyn JA, Yasuhara SE. Primary role of functional ischemia, quantitative evidence for the two-hit mechanism, and phosphodiesterase-5 inhibitor therapy in mouse muscular dystrophy. PLoS ONE. 2007; 2: e806.
- Ito K, Kimura S, Ozasa S, Matsukura M, Ikezawa M, Yoshioka K, et al. (2006). Smooth muscle-specific dystrophin expression improves aberrant vasoregulation in mdx mice. Hum Mol Genet. 2006; 15: 2266–2275.
- Loufrani L, Matrougui K, Gorny D, Duriez M, Blanc I, Lévy BI, et al. Flow (shear stress)-induced endothelium-dependent dilation is altered in mice lacking the gene encoding for dystrophin. Circulation. 2001; 103: 864–870.
- Dabiré H, Barthélémy I, Blanchard-Gutton N, Sambin L, Sampedrano CC, Gouni V, et al. Vascular endothelial dysfunction in Duchenne muscular dystrophy is restored by bradykinin through upregulation of eNOS and nNOS. Basic Res Cardiol. 107: 240.
- Nelson MD, Rader F, Tang X, Tavyev J, Nelson SF, Miceli MC. PDE5 inhibition alleviates functional muscle ischemia in boys with Duchenne muscular dystrophy. Neurology. 2014; 82: 2085-91.
- Harricane MC, Fabbrizio E, Lees D, Prades C, Travo P, Mornet D. Dystrophin does not influence regular cytoskeletal architecture but is required for contractile performance in smooth muscle aortic cells. Cell Biol Int. 1994; 18: 947-58.
- Morel JL, Dabertrand F, Fritz N, Henaff M, Mironneau J, Macrez N. The decrease of expression of ryanodine receptor sub-type 2 is reversed by gentamycin sulphate in vascular myocytes from mdx mice. J Cell Mol Med. 2009; 13: 3122–3130.
- De Stefano ME, Leone L, Lombardi L, Paggi P. Lack of dystrophin leads to the selective loss of superior cervical ganglion neurons projecting to muscular targets in genetically dystrophic mdx mice. Neurobiol Dis. 2005; 20: 929–942.
- Smith SA, Downey RM, Williamson JW, Mizuno M. Autonomic dysfunction in muscular dystrophy: a theoretical framework for muscle reflex involvement. Front Physiol. 2014; 5: 1–9.
- Schrimpf C, Teebken OE, Wilhelmi M, Duffield JS. The role of pericyte detachment in vascular rarefaction. J Vasc Res. 2014; 51: 247–258.
- Dunn JF, Tracey I, and Radda GK. Exercice metabolism in Duchenne Muscular Dystrophy: a biochemical and [31P]-nuclear magnetic resonance study of mdx mice. Proc R Soc Lond. 1993; 251: 201–206.
- Pant M, Sopariwala DH, Bal NC, Lowe J, Delfín DA, Rafael-Fortney J, et al. Metabolic dysfunction and altered mitochondrial dynamics in the utrophin-Dystrophin deficient mouse model of duchenne muscular dystrophy. PLoS ONE 2015; 10: 1–19.
- Rafael JA, Townsend ER, Squire SE, Potter AC, Chamberlain JS, Davies KE. Dystrophin and utrophin influence fiber type composition and post-synaptic membrane structure. Hum Mol Genet. 2000; 9: 1357– 1367.
- Rybalka E, Timpani CA, Cooke MB, Williams AD, Hayes A. Defects in Mitochondrial ATP Synthesis in Dystrophin-Deficient mdx Skeletal Muscles May Be Caused by Complex I Insufficiency. PLoS ONE. 2014; 9: e115763.