
Long-term window of ischemic tolerance: An evolutionarily conserved form of metabolic plasticity regulated by epigenetic modifications?
Nathalie Khoury, Kevin B. Koronowski and Miguel A. Perez-Pinzon*
Abstract
In the absence of effective neuroprotective agents in the clinic, ischemic and pharmacological preconditioning are gaining increased interest in the field of cerebral ischemia. Our lab recently reported that resveratrol preconditioning affords tolerance against a focal cerebral ischemic insult in mice that can last for at least 14 days in vivo making it the longest window of ischemic tolerance discovered to date by a single administration of a pharmacological agent. The mechanism behind this novel extended window of ischemic tolerance remains elusive. In the below commentary we discuss potential mechanisms that could explain this novel extended window of ischemic tolerance in the context of previously identified windows and the known mechanisms behind them. We also draw parallels from the fields of hibernation and hypoxia-tolerance, which are chronic adaptations to severe conditions of hypoxia and ischemia known to be mediated by a form of metabolic depression. We also briefly discuss the importance of epigenetic modifications in maintaining this depressed state of metabolism.
Preconditioning as an Approach to Reduce Brain Injury
Cerebral ischemia occurs in various forms such as stroke, cardiac arrest, subarachnoid hemorrhage and several neuro- and cardiac-surgeries1. Despite decades of pre-clinical and clinical research, cerebral ischemic injuries remain one of the leading causes of death and adult disability1. Numerous neuroprotective agents have been tested in the clinic yet nearly all have failed to promote significant recovery2. To date, a single FDA-approved drug for stroke exists, the thrombolytic agent recombinant tissue plasminogen activator (rtPA), yet it is only administered to a small percentage of stroke patients3,4. The failure of translation in cerebral ischemia research has been attributed at least in part to the rapid and complex cascade of events that ensue in the ischemic brain3,5 means to prepare the body against an ischemic injury prior to its onset6.
The brain responds naturally to lethal ischemia by rallying a host of molecular and cellular defenses that are deeply rooted in an organism’s genetic makeup7. Distinct from these mechanisms, the brain also responds to non-damaging ischemia, in anticipation of a lethal ischemic event to follow, by inducing evolutionarily conserved pathways that conjure some degree of protection7 (Figure 1). Many have drawn parallels to the likes of Nietzsche who stated “that which does not kill us, makes us stronger”8. While this was simply observation, the first scientific documentation of this phenomenon with regards to ischemia came in 1986 when Murry et al. discovered that in the heart, brief cycles of ischemia prior to a prolonged ischemic event actually lessens myocardial infarction9. In the same year, Schurr et al. reported that when rat hippocampal slices were pre-exposed to a short anoxic episode, they recovered from a subsequent longer ischemic insult while control slices did not10. Kitagawa et al. also showed in 1991 that brief interruption of carotid blood flow 2 days prior to global ischemia prevented CA1 pyramidal cell death in the gerbil hippocampus. Here, they coined the term ischemic tolerance11. The application of brief, non-damaging ischemia as a therapeutic intervention is now known as ischemic preconditioning (IPC) and has been demonstrated across species and tissues12,13. IPC has been extensively studied in rodent animal models as well as in in vitro cell models14,15. Interestingly, it was shown that numerous stimuli other than non-injurious ischemia can precondition the brain into a state of ischemic tolerance. These stimuli include hypoxia, hyperoxia, hypothermia, hyperthermia, inflammation, neurotoxins and many pharmacological agents6. Additionally, preconditioning with one stimuli can promote tolerance against an injurious dose of another stimuli, a phenomenon known as crosstolerance6. A promising pharmacological preconditioning agent that has been extensively studied by our group and others is resveratrol16-18, a natural polyphenol found on the skin of grapes, berries among other plants as well as in red wine19. Interestingly, resveratrol has not only shown promising results in models of cerebral ischemia, but it is currently in clinical trials for Alzheimer’s disease and have shown very promising preclinical results in other neurodegenerative as well as cardiovascular disorders19-21, which further underscores the importance of understanding its mechanism of action.
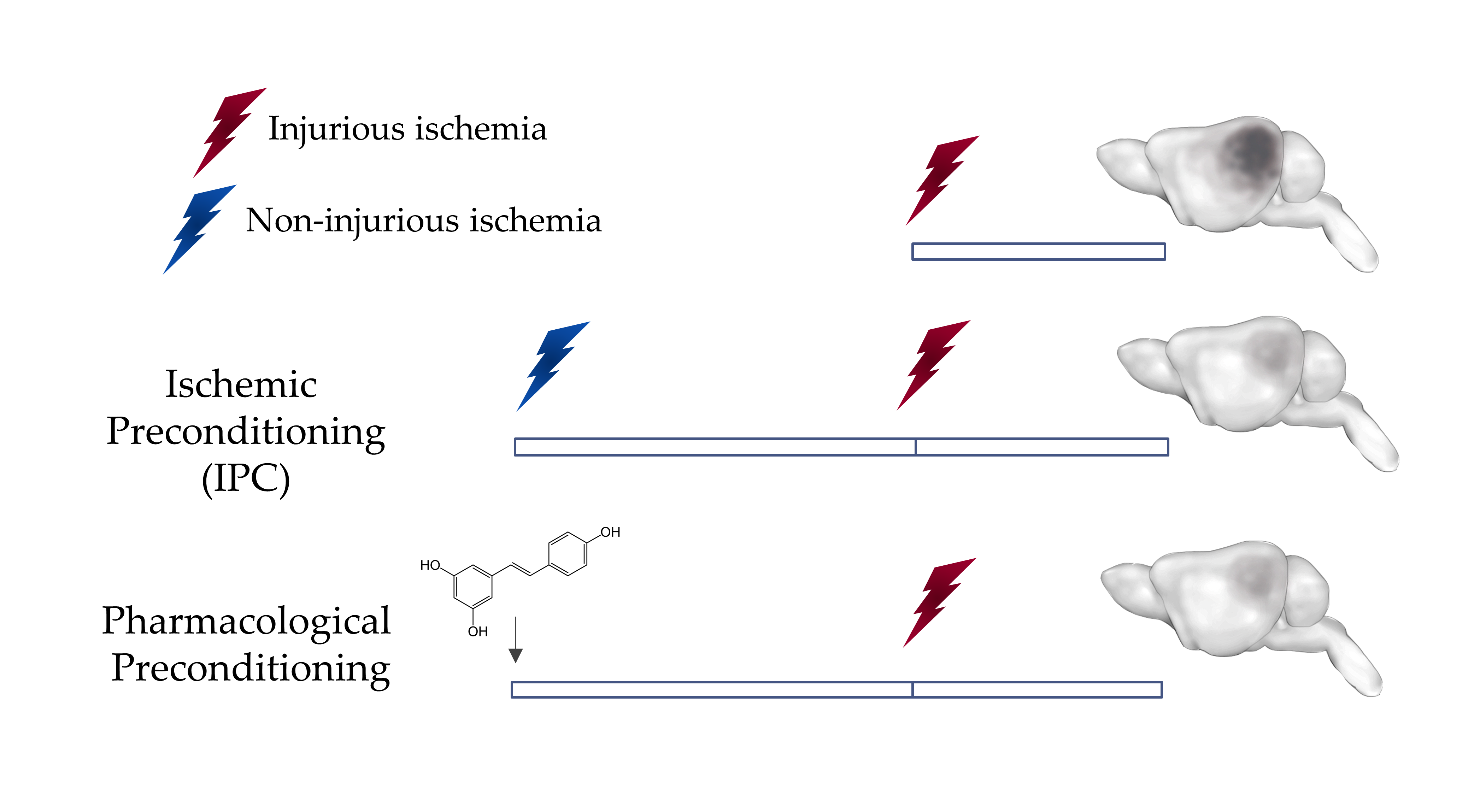
Figure 1: Preconditioning induces Ischemic Tolerance in the Brain
A non-injurious ischemic insult can protect the brain against a subsequent injurious ischemic insult. This phenomenon can be mimicked by the administration of some pharmacological agents such as resveratrol through a phenomenon known as pharmacological preconditioning.
Windows of Ichemic and Pharmacological Preconditioning
The ischemic tolerance mediated by preconditioning is observed within two transient windows22 (Figure 2). The first window, which is known as the rapid or short-term, appears minutes after preconditioning and lasts for a few hours. This window is thought to be mediated by posttranslational modifications to cellular components6,9,23,24. The second window, which is known as the delayed or long-term window, appears within a day after preconditioning and was thought to last for a maximum of 7 days after6,11. This window is known to be mediated by transcriptomic and epigenetic modifications as well as de novo protein synthesis22,25. Efforts have been made previously to extend the preconditioning window beyond 7 days. An interesting study revealed that repetitive hypoxic stimuli can actually extend the therapeutic window against cerebral ischemic for a remarkable period of 8 weeks in mice26. While repetitive hypoxia may lack translational value, IPC by way of remote limb preconditioning is a promising alternative for inducing ischemic tolerance27. Remote preconditioning is currently being clinically evaluated in the cardiac field28-30. Our lab has also made an effort to extend the preconditioning window. We previously showed that preconditioning with resveratrol (10 mg/kg), induces neuroprotection against middle cerebral artery occlusion (MCAo) in mice as well as against asphyxial cardiac arrest (ACA) in rats when administered two days prior to the insult14,16. Interestingly, we recently discovered that a single injection of resveratrol was sufficient to induce protection against an MCAO that lasts for at least 14 days in vivo, as shown by a reduction in infarct volume as well as an enhancement in the neurological score. This novel extended window of ischemic tolerance is currently the longest window of ischemic tolerance discovered to date by a single administration of pharmacological preconditioning31. Ongoing studies in our lab are aimed to determine the maximum time frame that a single resveratrol preconditioning (RPC) treatment can afford neuroprotection.
The fact that other studies in the field have not reported longer windows of ischemic tolerance via preconditioning could be explained by the nature of the preconditioning stimulus, its frequency of administration, and most notably its dose or intensity32. Higher doses of preconditioning agents do not necessarily increase the level of protection or its duration16,33. Studies in the field have reported an inverted U or J shape in response to preconditioning agents which highlights the importance of dose optimization in order to maximize adaptive responses and study mechanisms for potential clinical applications32.
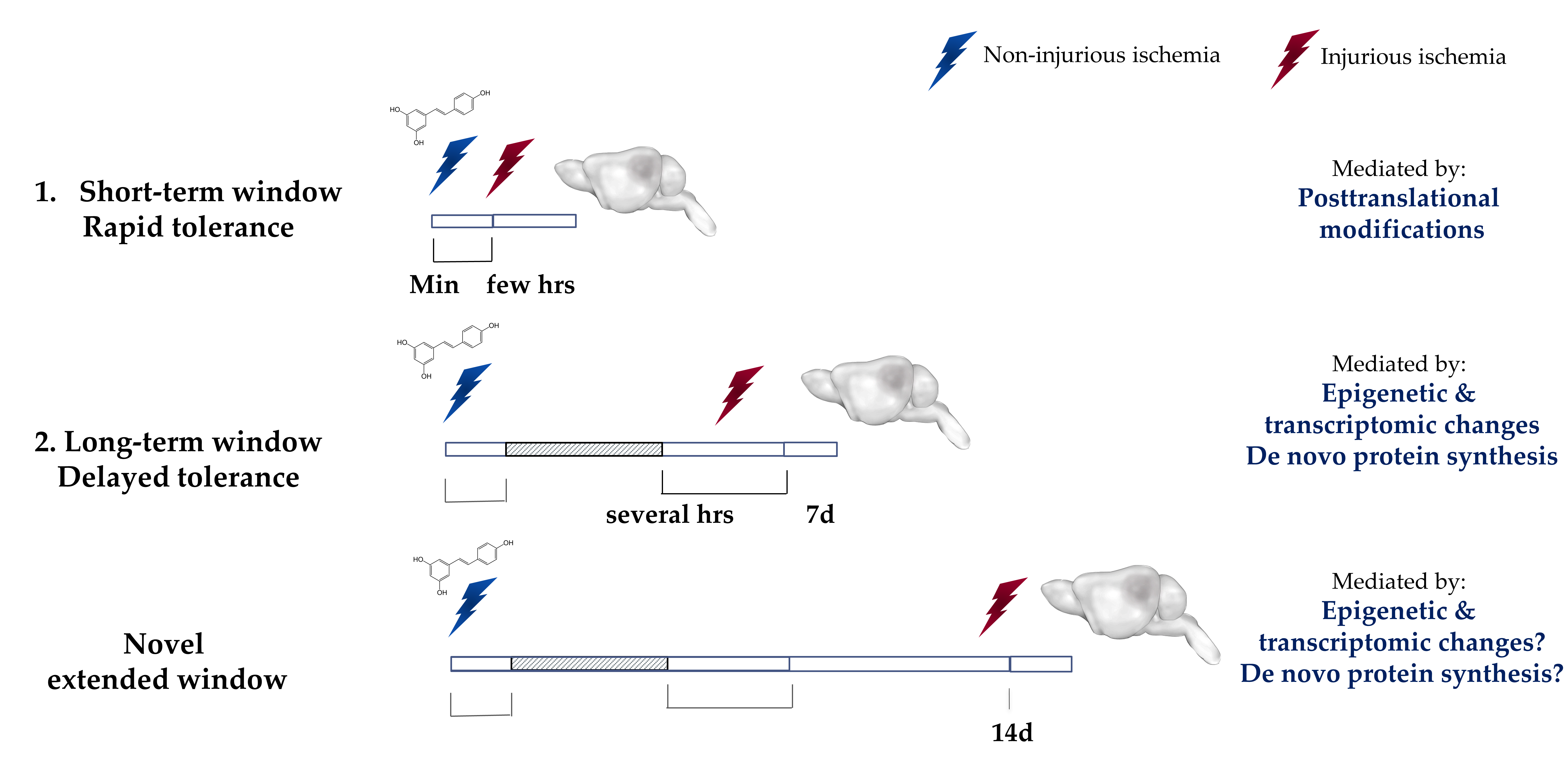
Figure 2: Ischemic Tolerance is observed within two transient time-windows.
The ischemic tolerance mediated by preconditioning is observed within two transient time-windows. The first window, known as the rapid or short-term, develops within minutes after the exposure and lasts for only a few hours after. This rapid protection is attributed to the transient post-translational modifications. The second window, known as the delayed or long-term, develops within days after the exposure but diminishes over the course of a week. The tolerance observed in the long-term window is attributed to epigenetic and transcriptomic modifications and de novo protein synthesis. Our laboratory has recently identified a novel extended window of ischemic tolerance that can last for at least 14 days in vivo after a single administration of resveratrol. Highlighted region represents a gap between the two windows where no protection has been observed. Min: minutes, hrs: hours, 7d: 7 days, 14d: 14 days.
Genomic Reprogramming in Ischemic Preconditioning
Knowing that resveratrol is an activator of the epigenetic enzyme SIRT1 (Sirtuin1), an NAD+-dependent histone deacetylase34, and taking into account the long-term therapeutic time-window observed, along with the extensive research supporting the importance of epigenetic modifications and transcriptomic changes in mediating the delayed window of ischemic tolerance35-39, we surmise that this extended long-term window afforded by RPC is mediated by genomic reprogramming events. Multiple epigenetic marks have been studied in the brain after ischemic preconditioning including DNA methylation40, histone acetylation41, histone methylation42 and histone phosphorylation43, among others35. So far the most promising and consistent results came from targeting histone deacetylases (HDACs) 5,44,45. Paradoxically, inhibting class I, IIa, IIb, and IV HDACs, which are zinc-dependent, showed promising results by decreasing infarct volume and enhancing behavioral outcome, while activating class III HDACs (sirtuins), which are NAD+-dependent, showed the same result35. Additional support to these observations came from the study which showed that zinc-dependent HDACs inhibitors and sirtuin activators can synergistically reduce the infarct volume as well as the neurological deficits41. Results from our lab also support a potential role for SIRT1 in mediating the observed tolerance within the long-term window of preconditioning. We have detected a significant upregulation in the protein levels of SIRT1 within the long-term window as well as increased binding to the promoter region of known mediators of preconditioning BDNF (brain-derived neurotrophic factors) and UCP2 (uncoupling protein 2) as revealed by chromatin immunoprecipitation31. Increased binding of SIRT1 to these promoters correlated with altered protein levels for BDNF and UCP2. Furthermore, the addition of sirtinol, a SIRT1 inhibitor, abolished the protection induced by RPC in organotypic hippocampal slices and in rodent animal models16,18. Ongoing studies in our lab will reveal more conclusive results about the importance of epigenetic modifications induced by SIRT1 in mediating this window of ischemic tolerance.
The first study to address the transcriptomic changes in the ischemic preconditioned brain was performed by Stenzel-Poore et al. in 2003. They reported a global downregulation in gene expression in the ischemic preconditioned brain m37,46. The study concluded that preconditioning reprograms the brain into a cellular state which resembles that seen during hibernation37,46. This cellular state of reduced metabolic activity is known to render the tissues and organs of hibernating animals resistant to hypoxic and hypoglycemic states experienced during the torpor phases of hibernation47,48. Subsequently, several other studies have aimed at identifying the transcriptomic changes in the ischemic preconditioned brain and have identified several common pathways which are misregulated after preconditioning including genes involved in excitotoxicity, apoptosis, inflammation, cytoskeleton remodeling, ribosome formation and ion channel activity, among others. Yet, an interesting observation common to many of these studies is a global downregulation of gene expression observed in the ischemic preconditioned and tolerant brain, reminiscent of the global downregulation in gene expression observed in hibernating and hypoxia-tolerant organisms7,22,49.
Metabolic Depression an Adaptation against Hypoxic and Ischemic Injuries
Metabolic depression, which can reach 1-20% of resting metabolic rate, is a conserved survival mechanism observed among many organisms across the animal kingdom. It is a common element observed among hibernation, torpor, estivation, anoxia, freeze tolerance, and diapause47,48. Animals enter this state when exposed to stressful environmental conditions such as extreme temperatures, oxygen limitation, food scarcity, or dehydration in order survive these unfavorable conditions over extended periods of time, ranging from weeks to months47,48. During the torpor phase of hibernation, the levels of oxygen consumption can be reduced to as low as 2 or 3% of basal rates, and heart rate reduced to an astonishing 3 to 10 beats per minutes50. Additionally, the rate of cerebral glucose consumption can reach 1-2% of active animals51, and cerebral blood flow is reduced as low as 10% of euthermic state52. Still, these animals tolerate the severe states of hypoxia and hypoglycemia for prolonged periods of time and recover from them with no noticeable damage to their organs. Such physiological conditions when experienced by humans, such as in the case of stroke, result in detrimental consequences and irreversible damages53. Researchers have been studying the mechanisms behind metabolic depression for decades in the hope of identifying endogenous adaptive mechanisms that could be induced in humans for possible therapeutic applications54. It is believed that inducing a less extreme “hibernation-like” state will protect stroke patients against the ischemia-mediated tissue damage54.
Hibernation falls on the extreme end of broad spectrum of phenotypes that involves metabolic depression as a form of energy conservation, yet other less extreme forms of metabolic plasticity are known to occur in other mammals. For example, slow-wave sleep which constitutes a milder form of metabolic depression is common to all mammals55. Thus metabolic plasticity exists among all mammals yet have different levels of flexibility55. The molecular basis of metabolic depression involves a controlled and coordinated suppression of ATP-consuming metabolic processes (such as transcription, translation, ATP-dependent ion pumps among others) as well as ATP generating processes so that a new homeostatic milieu is achieved that has a lower net rate of ATP turnover56. Additionally, the cells reprioritize the reduced ATP-availability to sustain vital cellular process such as increased antioxidant and chaperones to protect and maintain cellular components57.
The last decade or so has experienced an increased appreciation of the role of epigenetic modifications in maintaining cellular homeostasis in response to a changing environment58. The epigenetic machinery allows the cells to couple various intra- and extra- cellular signals to changes in gene expression in order to maintain a stable intracellular milieu58. Since metabolic depression is marked by a global suppression in the transcriptional and translational rates of cells over prolonged periods of time47,48, it is highly likely that these changes are maintained by reversible epigenetic modifications including DNA methylation, histone posttranslational modifications, and non-coding RNA such as microRNA, which are well known mechanisms that regulate transcriptional and translational rates within a cell58. Support to this hypothesis came from recent studies performed on the skeletal muscle and brown adipose tissue of the hibernating thirteen-lined squirrels59,60. These studies showed an increase in the expression and activity of histone deacetylases along with a reduction in the histone acetyl marks, which are known marks of active transcription, during the torpor phase of hibernation compared to the euthermic state thus supporting the reduced transcriptional rates observed during hibernation59,60. Similar observations were reported after prolonged anoxia in the freshwater turtle Trachemys scripta elegans which is also known to use metabolic depression as a survival mechanism to endure hypoxic conditions61,62.
Does preconditioning induce a depressed state of metabolism?
As mentioned previously the first study to assess the transcriptomic profile of preconditioned mouse brains reported a global suppression of gene expression after IPC specifically in genes involved in glucose metabolism, protein turnover, and ion channel abundance among others37,46. Consistent with their transcriptomic results, the authors also showed that preconditioning of cortical neuronal cultures reduces their whole-cell conductance as well as potassium-channel activity thus further supporting a depressed state of metabolic activtiy37,46. A subsequent study by Stapels et al. in 2010 showed that the transcriptional repressors known as the polycomb group proteins (PcG) are upregulated in the ischemic tolerant brain after IPC and are required to mediate the observed ischemic tolerance49. The authors showed that these PcG proteins associate with the promoter region of two potassium channels whose expression is reduced in the ischemic tolerant brain, and the knockdown or overexpression of these PcG protein in neuronal cultures increased or decreased the activity of voltage-gated potassium channels respectively49. The authors further show that the knockdown of these channels was sufficient to protect cells from ischemic injuries49. These studies support the role of metabolic depression regulated by epigenetic modifications as a mechanism behind ischemic tolerance seen after preconditioning. Additional support to the potential role of preconditioning in inducing a depressed state of metabolism comes from a study by Bracko et al. in 2014 who showed that ischemic tolerance seen in rats 3 days after preconditioning with 3-nitropropionic acid (NPA) correlates with a reduced state of metabolism in the brain as revealed by a significant reduction in the cerebral blood flow concomitant with a decrease in the brain’s energy charge potential as revealed by the abundance of the adenosine tri-, di-and mono-phosphates63. Another study has also reported a similar finding after preconditioning with cortical spreading depression and have attributed the observed ischemic tolerance to a reduction in the rate of the metabolism64. Furthermore, a recent study by Cui et al. in 2015 showed that repetitive exposure to hypoxia alters the proteomic profile of mouse brains causing a downregulating in the expression of genes involved in ATP (adenosine triphosphate) synthesis and citric acid cycle while upregulating those linked to glycolysis, which is similar to the adaptation used by hypoxia-tolerant organisms during their states of metabolic depression65. Moreover, several studies have showed that preconditioning reduces the brain’s energy expenditure by reducing its electrical activity as revealed by an alternation in the neurotransmitter signaling61. Preconditioned brains reduce their excitatory glutamate signaling while increasing the release of inhibitory neurotransmitters GABA and adenosine61. All these studies support a role for preconditioning in inducing a state of depressed metabolism in the brain which could explain the observed ischemic tolerance, whether resveratrol preconditioning also induces some form of metabolic plasticity in the brain within the long-term window of ischemic tolerance will be revealed by future studies in our lab.
Conclusion
The existence of a novel 14 day window of ischemic tolerance induced by resveratrol preconditoinng is reminescent of the long-term adaptations seen in nature in hiberanting and hypoxia-tolerant organisms against severe hypoxic and ischemic states. These adaptations might be evolutionary conserved at different levels across the animal kingdom and rely on a form of metabolic plasticity. Taking into account the importance of epigenetic machinery in mediating these adaptations, and in allowing a balanced homeostatic milieu in response to a continuously changing environment, we see the importance of understanding similar epigenetic changes in the context of ischemic tolerance and especially in the context of preconditoining. Understanding the molecular mechanisms behind this novel long-term window of ischemic tolerance might provide new insights into previously unexplored pathways and adaptations of ischemic tolerance.
References
- Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M. et al. Heart disease and stroke statistics--2015 update: a report from the American Heart Association. Circulation. 2015; 131: e29-322. Doi: 10.1161/CIR.0000000000000152.
- Kikuchi K, Tanaka E, Murai Y, Tancharoen S. Clinical trials in acute ischemic stroke. CNS Drugs. 2014; 28: 929-938. Doi: 10.1007/s40263-014-0199-6.
- Minnerup J, Sutherland BA, Buchan AM, Kleinschnitz C. Neuroprotection for stroke: current status and future perspectives. Int J Mol Sci. 2012; 13: 11753-11772. Doi: 10.3390/ijms130911753.
- Zivin JA. Acute stroke therapy with tissue plasminogen activator (tPA) since it was approved by the U.S. Food and Drug Administration (FDA). Ann Neurol. 2009; 66: 6-10. Doi: 10.1002/ana.21750.
- George PM, Steinberg GK. Novel Stroke Therapeutics: Unraveling Stroke Pathophysiology and Its Impact on Clinical Treatments. Neuron. 2015; 87: 297-309. Doi: 10.1016/j.neuron.2015.05.041.
- Stetler RA, Leak RK, Gan Y, Li P, Zhang F, Hu X. et al. Preconditioning provides neuroprotection in models of CNS disease: paradigms and clinical significance. Prog Neurobiol. 2014; 114: 58-83. Doi: 10.1016/j.pneurobio.2013.11.005.
- Cox-Limpens KE, Gavilanes AW, Zimmermann LJ, Vles JS. Endogenous brain protection: what the cerebral transcriptome teaches us. Brain Res. 2014; 1564: 85-100. Doi: 10.1016/j.brainres.2014.04.001.
- McDunn JE, Cobb JP. That which does not kill you makes you stronger: a molecular mechanism for preconditioning. Sci STKE 2005; 291: pe34. Doi: 10.1126/stke.2912005pe34.
- Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. 1986; Circulation. 74: 1124-1136.
- Schurr A, Reid KH, Tseng MT, West C, Rigor BM. Adaptation of adult brain tissue to anoxia and hypoxia in vitro. Brain res 1986; 374: 244-248.
- Kitagawa K, Matsumoto M, Kuwabara K, Tagaya M, Ohtsuki T, Hata R. et al. 'Ischemic tolerance' phenomenon detected in various brain regions. Brain res. 1991; 561: 203-211.
- Koti RS, Seifalian AM, Davidson BR. Protection of the liver by ischemic preconditioning: a review of mechanisms and clinical applications. Dig Surg. 2003; 20: 383-396.
- Wever KE, Menting TP, Rovers M, van der Vliet JA, Rongen GA, Masereeuw R. et al. Ischemic preconditioning in the animal kidney, a systematic review and meta-analysis. PLoS One. 2012; 7: e32296, Doi: 10.1371/journal.pone.0032296.
- Narayanan SV, Dave KR, Saul I, Perez-Pinzon MA. Resveratrol Preconditioning Protects Against Cerebral Ischemic Injury via Nuclear Erythroid 2-Related Factor 2. Stroke. 2015; 46: 1626-1632. Doi: 10.1161/STROKEAHA.115.008921.
- Morris-Blanco KC, Cohan CH, Neumann JT, Sick TJ, Perez-Pinzon MA. Protein kinase C epsilon regulates mitochondrial pools of Nampt and NAD following resveratrol and ischemic preconditioning in the rat cortex. J Cereb Blood Flow Metab. 2014; 34: 1024-1032. Doi: 10.1038/jcbfm.2014.51.
- Della-Morte D, Dave KR, DeFazio RA, Bao YC, Raval AP, Perez-Pinzon MA. et al. Resveratrol pretreatment protects rat brain from cerebral ischemic damage via a sirtuin 1-uncoupling protein 2 pathway. Neuroscience 2009; 159: 993-1002. Doi: 10.1016/j.neuroscience.2009.01.017.
- Singh N, Agrawal M, Doré S. Neuroprotective properties and mechanisms of resveratrol in in vitro and in vivo experimental cerebral stroke models. ACS Chem Neurosci. 2013; 4: 1151-1162. Doi:v10.1021/cn400094w.
- Raval AP, Dave KR, Pérez-Pinzón MA. Resveratrol mimics ischemic preconditioning in the brain. J Cereb Blood Flow Metab. 2006; 26: 1141-1147. Doi: 10.1038/sj.jcbfm.9600262.
- Tellone E, Galtieri A, Russo A, Giardina B, Ficarra S. Resveratrol: A Focus on Several Neurodegenerative Diseases. Oxid Med Cell Longev 2015; 392169. Doi: 10.1155/2015/392169.
- Petrovski G, Gurusamy N, Das DK. Resveratrol in cardiovascular health and disease. Ann N Y Acad Sci. 2011; 1215: 22-33. Doi: 10.1111/j.1749-6632.2010.05843.x.
- Turner RS, Thomas RG, Craft S, van Dyck CH, Mintzer J, Reynolds BA. et al. A randomized, double-blind, placebo-controlled trial of resveratrol for Alzheimer disease. Neurology. 2015; 85, 1383-1391, Doi: 10.1212/WNL.0000000000002035.
- Jimenez-Mateos, EM. Role of MicroRNAs in innate neuroprotection mechanisms due to preconditioning of the brain. Front Neurosci. 2015; 9: 118. Doi: 10.3389/fnins.2015.00118.
- Pérez-Pinzón MA, Xu GP, Dietrich WD, Rosenthal M, Sick TJ. Rapid preconditioning protects rats against ischemic neuronal damage after 3 but not 7 days of reperfusion following global cerebral ischemia. J Cereb Blood Flow Metab. 1997; 17: 175-182. Doi: 10.1097/00004647-199702000-00007.
- Pérez-Pinzón MA, Mumford PL, Rosenthal M, Sick TJ. Anoxic preconditioning in hippocampal slices: role of adenosine. Neuroscience. 1996; 75: 687-694.
- Durukan A, Tatlisumak T. Preconditioning-induced ischemic tolerance: a window into endogenous gearing for cerebroprotection. Exp Transl Stroke Med. 2010; 2: 2. Doi: 10.1186/2040-7378-2-2.
- Stowe AM, Altay T, Freie AB, Gidday JM. Repetitive hypoxia extends endogenous neurovascular protection for stroke. Ann Neurol. 2011; 69: 975-985. Doi: 10.1002/ana.22367.
- Meller R, Simon RP. A critical review of mechanisms regulating remote preconditioning-induced brain protection. J Appl Physiol. 2015; 119: 1135-1142. Doi: 10.1152/japplphysiol.00169.2015.
- Meybohm P, Bein B, Brosteanu O, Cremer J, Gruenewald M, Stoppe C. et al. A Multicenter Trial of Remote Ischemic Preconditioning for Heart Surgery. N Engl J Med 2015; 373: 1397-1407. Doi: 10.1056/NEJMoa1413579.
- Candilio L, Malik A, Ariti C, Barnard M, Di Salvo C, Lawrence D. et al. Effect of remote ischaemic preconditioning on clinical outcomes in patients undergoing cardiac bypass surgery: a randomised controlled clinical trial. Heart. 2015; 101: 185-192. Doi: 10.1136/heartjnl-2014-306178.
- White SK, Frohlich GM, Sado DM, Maestrini V, Fontana M, Treibel TA. et al. Remote ischemic conditioning reduces myocardial infarct size and edema in patients with ST-segment elevation myocardial infarction. JACC Cardiovasc Interv. 2015; 8: 178-188. Doi: 10.1016/j.jcin.2014.05.015.
- Koronowski KB, Dave KR, Saul I, Camarena V, Thompson JW, Neumann JT. et al. Resveratrol Preconditioning Induces a Novel Extended Window of Ischemic Tolerance in the Mouse Brain. Stroke. 2015; 46: 2293-2298. Doi: 10.1161/STROKEAHA.115.009876.
- Calabrese, E. J. Preconditioning is hormesis part I: Documentation, dose-response features and mechanistic foundations. Pharmacol Res. 2016; Doi: 10.1016/j.phrs.2015.12.021.
- Hermann DM, Zechariah A, Kaltwasser B, Bosche B, Caglayan AB, Kilic E. et al. Sustained neurological recovery induced by resveratrol is associated with angioneurogenesis rather than neuroprotection after focal cerebral ischemia. Neurobiol Dis. 2015; 83: 16-25. Doi: 10.1016/j.nbd.2015.08.018.
- Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG. et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature. 2003; 425: 191-196. Doi: 10.1038/nature01960.
- Thompson JW, Dave KR, Young JI, Perez-Pinzon MA. Ischemic preconditioning alters the epigenetic profile of the brain from ischemic intolerance to ischemic tolerance. Neurotherapeutics. 2013; 10: 789-797. Doi: 10.1007/s13311-013-0202-9.
- Chisholm NC, Henderson ML, Selvamani A, Park MJ, Dindot S, Miranda RC. et al. Histone methylation patterns in astrocytes are influenced by age following ischemia. Epigenetics. 2015; 10: 142-152. Doi: 10.1080/15592294.2014.1001219.
- Stenzel-Poore MP, Stevens SL, Simon RP. Genomics of preconditioning. Stroke. 2004; 35: 2683-2686. Doi: 10.1161/01.STR.0000143735.89281.bb.
- Stenzel-Poore MP, Stevens SL, King JS, Simon RP. Preconditioning reprograms the response to ischemic injury and primes the emergence of unique endogenous neuroprotective phenotypes: a speculative synthesis. Stroke. 2007; 38: 680-685. Doi: 10.1161/01.STR.0000251444.56487.4c.
- Tang Y, Pacary E, Fréret T, Divoux D, Petit E, Schumann-Bard P. et al. Effect of hypoxic preconditioning on brain genomic response before and following ischemia in the adult mouse: identification of potential neuroprotective candidates for stroke. Neurobiol Dis. 2006; 21: 18-28. Doi:10.1016/j.nbd.2005.06.002.
- Endres M, Fan G, Meisel A, Dirnagl U, Jaenisch R. Effects of cerebral ischemia in mice lacking DNA methyltransferase 1 in post-mitotic neurons. Neuroreport. 2011; 12: 3763-3766.
- Lanzillotta A, Pignataro G, Branca C, Cuomo O, Sarnico I, Benarese M. et al. Targeted acetylation of NF-kappaB/RelA and histones by epigenetic drugs reduces post-ischemic brain injury in mice with an extended therapeutic window. Neurobiol Dis. 2013; 49: 177-189. Doi: 10.1016/j.nbd.2012.08.018.
- Mudie S, Bandarra D, Batie M, Biddlestone J, Moniz S, Ortmann B. et al. PITX1, a specificity determinant in the HIF-1alpha-mediated transcriptional response to hypoxia. Cell Cycle. 2014; 13: 3878-3891. Doi: 10.4161/15384101.2014.972889.
- Crowe SL, Tsukerman S, Gale K, Jorgensen TJ, Kondratyev AD. Phosphorylation of histone H2A.X as an early marker of neuronal endangerment following seizures in the adult rat brain. J Neurosci. 2011; 31: 7648-7656. Doi: 10.1523/JNEUROSCI.0092-11.2011.
- Aune SE, Herr DJ, Kutz CJ, Menick DR. Histone Deacetylases Exert Class-Specific Roles in Conditioning the Brain and Heart Against Acute Ischemic Injury. Front Neurol. 2015; 6: 145. Doi: 10.3389/fneur.2015.00145.
- Baltan S, Morrison RS, Murphy SP. Novel protective effects of histone deacetylase inhibition on stroke and white matter ischemic injury. Neurotherapeutics. 2013; 10: 798-807. Doi: 10.1007/s13311-013-0201-x.
- Stenzel-Poore MP, Stevens SL, Xiong Z, Lessov NS, Harrington CA, Mori M. et al. Effect of ischaemic preconditioning on genomic response to cerebral ischaemia: similarity to neuroprotective strategies in hibernation and hypoxia-tolerant states. Lancet. 2003; 362: 1028-1037. Doi: 10.1016/S0140-6736(03)14412-1.
- Storey KB. Regulation of hypometabolism: insights into epigenetic controls. J Exp Biol. 2015; 218: 150-159. Doi: 10.1242/jeb.106369.
- Biggar KK, Storey KB. The emerging roles of microRNAs in the molecular responses of metabolic rate depression. J Mol Cell Biol. 2011; 3: 167-175. Doi: 10.1093/jmcb/mjq045.
- Stapels M, Piper C, Yang T, Li M, Stowell C, Xiong ZG. et al. Polycomb group proteins as epigenetic mediators of neuroprotection in ischemic tolerance. Sci Signal. 2010; 3: ra15. Doi: 10.1126/scisignal.2000502.
- Hampton M, Nelson BT, Andrews MT. Circulation and metabolic rates in a natural hibernator: an integrative physiological model. Am J Physiol Regul Integr Comp Physiol. 2010; 299: R1478-1488. Doi: 10.1152/ajpregu.00273.2010.
- Frerichs KU, Dienel GA, Cruz NF, Sokoloff L, Hallenbeck JM. Rates of glucose utilization in brain of active and hibernating ground squirrels. Am J Physiol. 1995; 268: R445-453.
- Dave KR, Christian SL, Perez-Pinzon MA, Drew KL. Neuroprotection: lessons from hibernators. Comp Biochem Physiol B Biochem Mol Biol. 2012; 162: 1-9. Doi: 10.1016/j.cbpb.2012.01.008.
- Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999; 22: 391-397.
- Forreider B, Pozivilko D, Kawaji Q, Geng X, Ding Y. Hibernation-like neuroprotection in stroke by attenuating brain metabolic dysfunction. Prog Neurobiol. 2016; Doi: 10.1016/j.pneurobio.2016.03.002.
- van Breukelen F, Martin SL. The Hibernation Continuum: Physiological and Molecular Aspects of Metabolic Plasticity in Mammals. Physiology (Bethesda). 2015; 30: 273-281. Doi: 10.1152/physiol.00010.2015.
- Storey KB. Anoxia tolerance in turtles: metabolic regulation and gene expression. Comp Biochem Physiol A Mol Integr Physiol. 2007; 147: 263-276. Doi: 10.1016/j.cbpa.2006.03.019.
- Wu CW, Biggar KK, Storey KB. Biochemical adaptations of mammalian hibernation: exploring squirrels as a perspective model for naturally induced reversible insulin resistance. Braz J Med Biol Res 2013; 46: 1-13.
- Colvis CM, Pollock JD, Goodman RH, Impey S, Dunn J, Mandel G. et al. Epigenetic mechanisms and gene networks in the nervous system. J Neurosci. 2005; 25: 10379-10389. Doi: 10.1523/JNEUROSCI.4119-05.2005.
- Morin P Jr, Storey KB. Evidence for a reduced transcriptional state during hibernation in ground squirrels. Cryobiology. 2006; 53: 310-318. Doi: 10.1016/j.cryobiol.2006.08.002.
- Biggar Y, Storey KB. Global DNA modifications suppress transcription in brown adipose tissue during hibernation. Cryobiology. 2014; 69: 333-338. Doi: 10.1016/j.cryobiol.2014.08.008.
- Perez-Pinzon MA. Mechanisms of neuroprotection during ischemic preconditioning: lessons from anoxic tolerance. Comp Biochem Physiol A Mol Integr Physiol. 2007; 147: 291-299. Doi: 10.1016/j.cbpa.2006.08.032.
- Krivoruchko A, Storey KB. Epigenetics in anoxia tolerance: a role for histone deacetylases. Molecular and cellular biochemistry. 2010; 342: 151-161. Doi: 10.1007/s11010-010-0479-5.
- Bracko O, Di Pietro V, Lazzarino G, Amorini AM, Tavazzi B, Artmann J. et al. 3-Nitropropionic acid-induced ischemia tolerance in the rat brain is mediated by reduced metabolic activity and cerebral blood flow. J Cereb Blood Flow Metab. 2014; 34: 1522-1530. Doi: 10.1038/jcbfm.2014.112.
- Otori T, Greenberg JH, Welsh FA. Cortical spreading depression causes a long-lasting decrease in cerebral blood flow and induces tolerance to permanent focal ischemia in rat brain. J Cereb Blood Flow Metab. 2003; 23: 43-50.
- Cui C, Zhou T, Li J, Wang H, Li X, Xiong J. et al. Proteomic analysis of the mouse brain after repetitive exposure to hypoxia. Chem Biol Interact. 2015; 236: 57-66. Doi: 10.1016/j.cbi.2015.04.010.