
TrkB and the calpain dependent-Tc TrkB receptor isoforms: possible neurological therapeutic targets
Víctor Danelon1, Andrea B. Cragnolini1, Daniel Masco1*
Abstract
Several neurological conditions share a characteristic feature: an increase in intracellular calcium levels ([Ca2+]i). It has been demonstrated that calcium influx induces changes ranging from an increase in the expression levels of several genes to the activation of proteases such as calpains. Calpains are a family of Ca2+-dependent non-lysosomal cysteine proteases, whose substrates include several proteins that play critical roles in several cellular functions including synaptic plasticity and neuronal apoptosis.
TrkB is a type of tyrosine related kinase receptor that can promote neuronal survival and differentiation upon ligand binding. It has been recently shown that in several neurological diseases, the level of full-length TrkB protein decreases before the onset of neuronal death due to one of two different processes: a) a reverse regulation of TrkB isoforms mRNA, or b) calpain-mediated processing of TrkB full-length, which yields a truncated form of TrkB (Tc-TrkB). Because the most notorious feature of calpain proteolytic activity is that the products of calpain-mediated cleavage may have biological activity, here we review the hypotheses by which calpain-generated isoform Tc-TrkB may perform biological functions.
Abbreviations:
BDNF: Brain Derived Neurotrophic Factor; Cdk5 (Cyclin-Dependent Kinase 5); CNS: Central Nervous System; Erk/MAPK: extracellular related kinase.
ICD: intracellular fragment; NCX: Na+–Ca2+ exchanger; NMDAr: N-Methyl-D-Aspartate receptor; NT: neurotrophins; NT4: Neurotrophin 4; p75NTR: 75 neurotrophin receptor; PI3K: Phosphoinositide 3-kinase; PKC: protein kinase C; PKM: protein kinase M; PLC: phospholipase c; proBDNF: BDNF immature isoform; PSD: Postsynaptic Density Protein; SE: status epilepticus; Tc-TrkB: calpain-truncated TrkB receptor; TrkB-Tk: truncated TrkB receptor; TrkB: full-length Tropomyosin related Kinase B Receptor; TrkB-ICD: intracellular fragment of TrkB;
Introduction
Neurotrophins (NT) are a set of proteins that act through high affinity receptors found on the cell surface. Over the past two decades, there has been a major increase in NT research studies, especially in the Central Nervous System (CNS)9,10,25,27. NT play a critical role not only in the development and maintenance of the nervous system, but also in neuropathological disorders. It has been suggested that endogenous changes in NT expression can contribute to the development of neurological conditions. To study NT function is challenging due to the different biological outcomes they elicit, which range from trophic, survival, or neuritogenic differentiation to, putatively, neuronal death 9,10,22,25,27,42,43. The NT, more precisely BDNF (Brain Derived Neurotrophic Factor), triggers signal transduction by binding to TrkB (Tropomyosin receptor kinase B)28 and p75NTR (p 75 neurotrophins receptor) receptors.
Several TrkB receptor isoforms have been described, and are abundantly expressed in the brain: a full-length TrkB (TrkB), and the truncated isoforms TrkB-Tk. TrkB activation trigger different intracellular signaling cascades, including mitogen-activated protein kinase/extracellular signal regulated protein kinase (MAPK/ERK), the phospholipase C-γ (PLCγ), and the phosphoinositol-3 kinase (PI3K)/protein kinase B (AKT) pathways, which in turn induce cellular processes such as differentiation, proliferation and survival in different cell types9,10. Moreover, these physiological processes are regulated by different mechanisms, such as spatial and temporal changes in the level of TrkB ligands, the ratio between TrkB and TrkB-Tk, intracellular signaling pathways, and gene expression. Despite the fact that the biological role of TrkB-Tk receptor isoforms is not yet fully known, it has been recently associated with several functions in neurons and glial cells, such as the regulation of BDNF levels at the extracellular site, the activation of intracellular signaling pathways, morphological changes in neurons and glial cells, and dominant-negative inhibition of TrkB signaling. Since the function and signaling of TrkB-Tk receptor has been described in detail in excellent reviews6,14,23,34,36, they are beyond the scope of this revision.
Together TrkB and the described isoforms have recently been associated with several neurological conditions that affect the CNS, such as ischemia, Alzheimer’s disease (AD), and status epilepticus (SE), where excitotoxic insults disrupt the balance among TrkB/TrkB Tk/p75NTR ratio and the increase in the synthesis and release of BDNF/proBDNF are prior events to the onset of neuronal death19,24,40,42,43,44,46. On one hand, this imbalance produces a decrease at the level of both TrkB transcripts and protein. On the other hand, evidence indicates that TrkB-Tk and p75NTR receptor levels increase due to upregulated at the transcriptional and translational levels, since inhibitors of both processes prevent the excitotoxic-induced upregulation observed43,44,46. Thus, it can be inferred that trkb mRNA splicing mechanisms may change under excitotoxic conditions. Even though there are increasing evidences supporting these findings, the biological consequences of the TrkB/TrkB-Tk imbalance is still a matter of debate19,24,40,44,46.
Also it has been demonstrated that BDNF/proBDNF (BDNF immature isoform) have key roles in determine neuronal death once the pathology has occurred. In vivo results from our lab show when intra-hippocampal infusion of function-blocking antibodies against BDNF avoided the down-regulation of TrkB and prevent neuronal death following the SE insult. In line with this, the infusion of BDNF protein after SE exacerbated neuronal degeneration and surprisingly, inhibiting BDNF protein synthesis, due to the infusion of antisense oligonucleotides, induced a rapid decrease of BDNF protein levels, and SE-induced neuronal damage was prevented42. These results show that there is a complex interplay between the NT and their receptors before the onset of neuronal death.
Calpain protease mediates the TrkB receptor degradation following an excitotoxic insult
Neuronal depolarization induces an increase in intracellular calcium levels ([Ca2+]i), which exerts a variety of functions, ranging from increased expression of several genes to the activation of proteases such as calpains18,21,32,35. Calpains constitute a family of Ca2+-dependent non-lysosomal cysteine proteases that play key roles in various cellular functions, including synaptic plasticity and neuronal apoptosis. To achieve its functions, calpain acts on different substrates, including cytoskeletal proteins (e.g. α-spectrin, neurofilaments), membrane receptors (e.g. NMDA receptor), and other proteases. When active, calpain modifies the structure and activity of their targets by limited proteolysis18.
Calpains have two modes of action depending on the [Ca2+]i found in the cytoplasm: in a physiological way and in pathological conditions, where there is calcium overload mainly by over-activation of the NMDA receptor (NMDAr), resulting in calpain overactivation11,32. It has been recently observed that, in ischemia, AD, and experimental SE models, a drop in TrkB level is not only due to a decrease at the transcriptional level. A TrkB-calpain-dependent-processing is another mechanism that cooperates with the imbalance observed12,24,44,46. This proteolityc cleavage results in a truncated protein, Tc-TrkB, which lacks the tyrosine-kinase domain, and it is comparable to the synthesized TrkB-Tk isoform (first demonstrated by Vidaurre et al.44). Jeronimo-Santos et al.24has confirmed these results, and established that the TrkB site for calpain-cleavage is located between the Shc-binding site (Tyr515) and the TrkB kinase domain. Consequently, the Tc-TrkB receptor contains the Shc-binding site, whereas the remaining intracellular fragment (TrkB-ICD) contains the complete tyrosine kinase domain of TrkB receptor. These data indicate that the proteolytic processing triggered by calpain generates two TrkB-derived proteins.
Tc-TrkB increases in the dendritic and axonal domains before the onset of neuronal death.
Degeneration of neuritic processes in neurons is an early event in a wide range of neurodegenerative diseases as well as in other neurological conditions4,35,48. We found that the neurotrophin receptor TrkB is proteolitically processed by calpain in the dendritic and axonal domains of neurons committed to die, and this constitutes an early step in the neuronal degenerative process12. While the TrkB level decreases, the Tc-TrkB concurrently increases, leading to a TrkB/Tc-TrkB imbalance, which we believe it is linked to neurodegeneration. When a specific calpain activity blocker agent was used, the TrkB/Tc-TrkB imbalance was prevented, the neuronal processes were protected from degeneration and an enhanced neuronal survival was observed. Additionally, ANA-12, a specific TrkB antagonist7, blocked SE-induced neuronal death. By the time that ANA-12 was added to the neurons in culture, a significant decrease in the level of TrkB had already occurred. Thus, we believe that the observed effect of the ANA-12 antagonist did not result from TrkB inhibition, but mostly from an effect on Tc-TrkB. Others have shown an increase in the synthesis of TrkB-Tk receptor following excitotoxic stimulation, which takes place prior to the onset of neuronal death24,44. However, the possibility that neuronal death is prevented by ANA-12 through inhibition of signaling pathways downstream TrkB-Tk can be discarded, since blocking TrkB-Tk protein synthesis did not affect neuronal death44. It will be important to further investigate the biological role of Tc-TrkB in this process.
Tc-TrkB may acquire a biological function following calpain-dependent TrkB processing
Several studies show that some proteolytic products of calpain-mediated cleavage carry biological activity18. A well-known example of a calpain substrate proteolytic product with biological activity is the p35 protein, a neuron-specific activator of cyclin-dependent kinase 5 (CDK5). When calpain is activated, p35 cleavage results in p25, which steadily activates CDK5, and p10 proteins, also biologically active30. This cyclin-dependent kinase has been implicated in several biological functions, including modulation of synaptic plasticity and hyper-phosphorylation of Tau protein, an AD hallmark15,30,38. Similarly to the dual functions of CDK5, the metabotropic glutamate receptor mGluR1α has also the potential to be neuroprotective as well as to contribute to neurotoxicity. In an excitotoxic model, calpain mediate the proteolityc cleavage of the C-terminal tail of mGluR1α which impair its capability to induce neuroprotective effects via Akt activation. The cleavage of mGluR1α helps to maintain a positive feedback regulation in excitotoxicity (involving calpain and mGluR1α), which results in a pathological intracellular calcium increased47. In line with the latter, another calpain target includes the NCX3 subtype of the Na+-Ca2+ exchanger (NCX) whose proteolytic inactivation helps to maintain high cytoplasmic calcium levels3.
Several studies have shown that calpain-processed proteins have effects at the synaptic level by modulating burst firing and by modifying the localization of receptors in normal and pathological conditions. Calpain cleaves protein kinase C (PKC) which releases the active protein kinase M fragment (PKM) which can modulate the development and maintenance of burst firing, a process that enhances neurotransmitter release to the synaptic cleft31. The postsynaptic density protein 95 (PSD-95), a scaffold protein that clusters several receptors together, is also a calpain target45. When the PSD-95 is cleaved, the anchoring of NMDAR is modified13,33. In a Huntington disease model, calpain is involved in the re-localization of membrane receptors. In this pathological condition, calpain cleaves the C-terminal GluN2B subunits, resulting in the relocation of neuroprotective synaptic NMDA receptors to extrasynaptic sites16,17. Moreover, the Huntingtin protein (Htt) fragments cleavaged by calpain can translocate to the nucleus, with toxic effects on the cell20. In addition, it is known that cleavage of procaspase-3 by calpain leads to activation of caspase, which can degrade the calpain specific endogenous inhibitor Calpastatin, which in turn accelerates calpain activation5,48.
Furthermore, as mentioned above, the calpain-processed TrkB generates TrkB-ICD24,40. It is feasible to think that this fragment might also acquire biological functions, similarly to what happens to the above mentioned calpain-products. Other authors have found that many tyrosine kinase receptors can experience proteolytic cleavage by γ-secretases, caspases, and/or metalloproteases, generating ICD fragments with cellular functions1,40. Moreover, the neurotrophin receptor p75NTR undergoes a rapid α- and γ-secretase-dependent cleavage, following neurotrophin-mediated Trk activation. Therefore, p75NTR receptor cleavage releases ICD26, which facilitates NT-Trk-dependent Akt activation8.
Based on all the information supporting a functional role of calpain-cleaved products, it would not be surprising to find that the TrkB-calpain-cleaved products can perform a biological function.
Therapeutic targeting of the TrkB receptor
In this article, after reviewing the latest knowledge of TrkB processing by calpain, we discuss the rationale underlying the selective targeting of calpain to prevent the Tc-TrkB mediated neuronal death. A good strategy has to be proposed in order to prevent the axonal and dendritic domain degeneration once the pathology has occurred. Among them to be considered is the intraperitoneal administration (i.p.) of a calpain activity blocker, as was already been demonstrated by Araujo et al.2. Using kainic acid (KA) as an in vivo model of SE, Araujo et al. demonstrated that i.p. infusion of MDL 28170, a calpain activity blocker that pass through the blood-brain barrier, avoided the extent of the lesion in the CA1 area in the hippocampus of KA-injected rats. Whether the i.p. administration of the calpain activity blocker prevents the increase in the Tc-TrkB in the axonal and dendritic domain and thus, explain the decrease in the neurodegeneration, is a good hypothesis to be tested in order to develop new pharmacological treatment once the pathology has occurred.
Another plausible therapeutic strategy to prevent neurodegeneration would be the inhibition of the BDNF-protein synthesis. We already showed that this approach markedly decrease neuronal death after SE in vivo42. In line with this, others have shown that inhibiting the BDNF-TrkB complex it is associated with a decreased in the epileptogenesis in SE models 37. New strategies to avoid neurodegeneration can be focus on TrkB processing with the aim of developing promising strategies to treat SE patients.
Concluding remarks
Recently published results indicate that physiological or pathological increase in [Ca2+]I induces an increase in calpain activation, which, in turn, leads to the proteolytic cleavage of several substrates. The excitotoxic insult selectively upregulates TrkB-Tk protein level. Also, it simultaneously decreases TrkB level via transcriptional deregulation and by increasing TrkB calpain-proteolityc cleavage. This process results in two new isoforms: Tc-TrkB and TrkB-ICD. Further experiments are needed to elucidate whether both calpain-dependent-TrkB-cleaved products may acquire new biological functions.
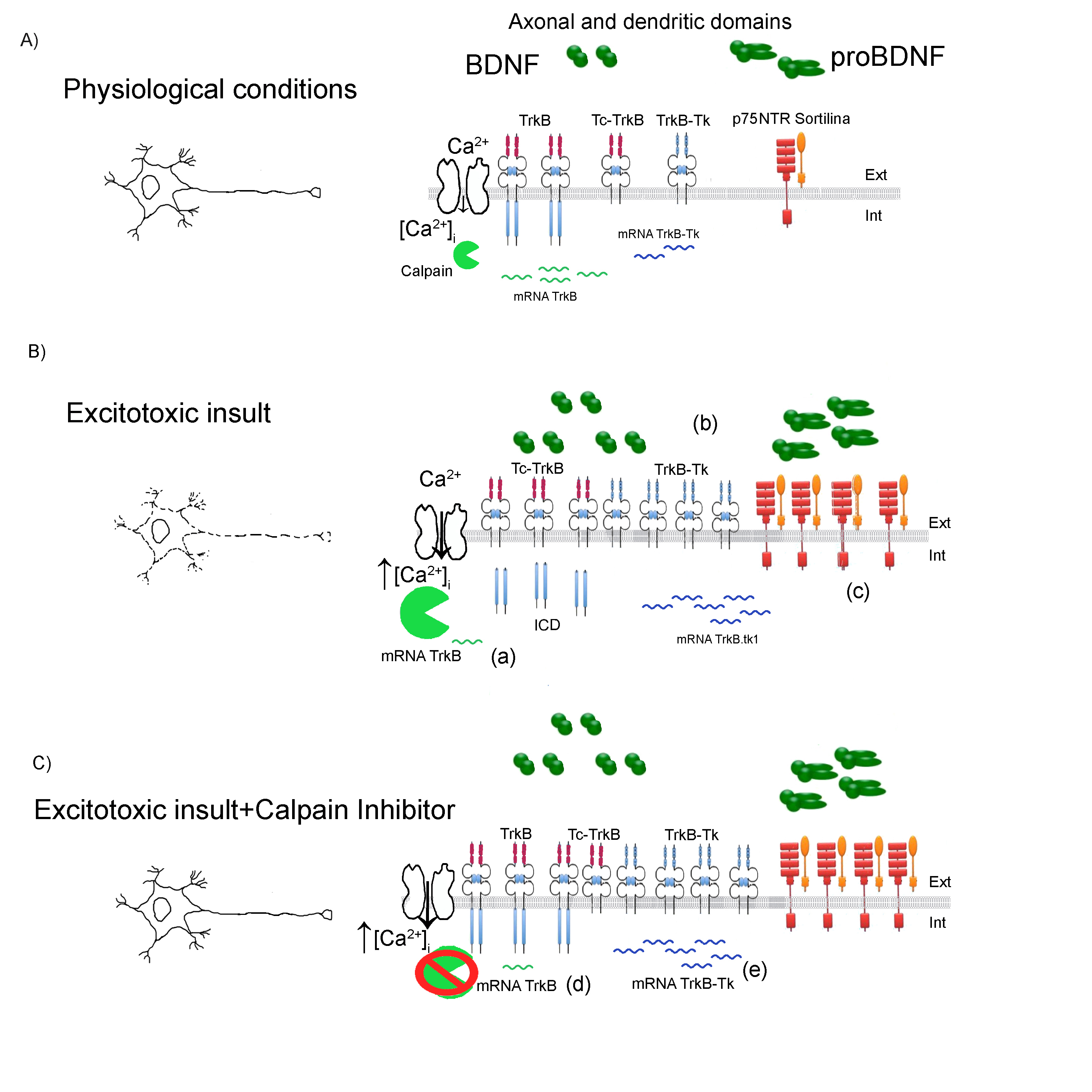
Figure 1: Schematic representation model of TrkB /Tc-TrkB/TrkB-Tk dysregulation after an excitotoxic insult.
A) Physiological condition. Neuronal depolarization induces an increase in the intracellular calcium levels ([Ca2+]i) which participate in several functions: from increasing the expression of several genes (among them is the trkb gene) to the activation of proteases such as calpains. B) Excitotoxic insult. An excitotoxic insult induces a neuronal hyper-depolarization which produce a massive increase in the ([Ca2+]i). In these conditions, calpain proteases experience a pathological activation that cleaves several substrates, including the TrkB receptor. This proteolityc cleavage induces a decrease in TrkB levels and increase in the Tc-TrkB. Also, another mechanism that contributes to the decrease in the TrkB protein levels is the change in the splicing mechanisms of the TrkB transcripts: increasing the TrkB-Tk mRNA levels and decreasing the TrkB mRNA levels. (a) In Jerónimo-Santos et al24, there is no change in the TrkB mRNA levels following amyloid-β peptide treatment. In Unsain et al.43 there is also an increase in the levels of proBDNF, BDNF and p75NTR following SE (b) and (c). C) Excitotoxic insult+Calpain inhibitor. Although there is an increase in the [Ca2+]I, when calpain inhibitor is added prior to, during and after the excitotoxic insult, a complete neuroprotection is observed 12. Xie et al.46 shows that when a calpain inhibitor is added to neuronal cultures there is an increase in the TrkB mRNA (d) and a decrease in the TrkB-Tk mRNA (e). Ext: Extracellular; Int: intracellular compartment.
References
- Ancot F, Foveau B, Lefebvre J, Leroy C, Tulasne D. Proteolytic cleavages give receptor tyrosine kinases the gift of ubiquity. Oncogene. 2009; 28:2185–2195.
- Araújo IM, Gil JM, Carreira BP, Mohapel P, Petersen A, Pinheiro PS, et al. Calpain activation is involved in early caspase-independent neurodegeneration in the hippocampus following status epilepticus. J Neurochem. 2008; 105(3):666-76
- Bano D, Young KW, Guerin CJ, Lefeuvre R, Rothwell NJ, Naldini L, et al. Cleavage of the plasma membrane Na+/Ca2+ exchanger in excitotoxicity. Cell. 2005; 120 (2): 275–285
- Bevers MB, Neumar RW. Mechanistic role of calpains in postischemic neurodegeneration. J. Cereb. Blood Flow Metab. 2008; 28 (4): 655–673.
- Blomgren K, Zhu C, Wang X, Karlsson JO, Leverin AL, Bahr BA, et al. (2001) Synergistic activation of caspase-3 by m-calpain after neonatal hypoxia–ischemia: a mechanism of “pathological apoptosis”? J. Biol. Chem. 2001; 276 (13): 10191–10198.
- Carim-Todd L, Bath KG, Fulgenzi G, Yanpallewar S, Jing D, Barrick CA. et al. Endogenous Truncated TrkB.T1 Receptor Regulates Neuronal Complexity and TrkB Kinase Receptor Function In Vivo. The Journal of Neuroscience. 2009; 29(3):678–685
- Cazorla M, Prémont J, Mann A, Girard N, Kellendonk C, Rognan D. Identification of a low-molecular weight TrkB antagonist with anxiolytic and antidepressant activity in mice. J Clin Invest. 2011; 121 (5): 1846–1857.
- Ceni C, Kommaddi RP, Thomas R, Vereker E, Liu X, McPherson PS, et al. The p75NTR intracellular domain generated by neurotrophin-induced receptor cleavage potentiates Trk signaling. J Cell Sci. 2010; 123 (Pt 13):2299-307.
- Chao MV. Neurotrophins and their receptors: a convergence point for many signaling pathways. Nat Rev Neurosci. 2003; 4: 299-309.
- Chao M. Neurotrophins: To cleave or not to cleave. Neuron. 2002; 33(1):9-12.
- Curcio M, Salazar IL, Mele M, Canzoniero LM, Duarte CB. Calpains and neuronal damage in the ischemic brain: The swiss knife in synaptic injury. Prog Neurobiol. 2016; 143:1-35.
- Danelon V, Montroull LE, Unsain N, Barker PA, Mascó DH. Calpain-dependent truncated form of TrkB-FL increases in neurodegenerative processes. Mol Cell Neurosci. 2016; 75:81-92.
- Dong YN, Waxman EA, Lynch DR. Interactions of postsynaptic density-95 and the NMDA receptor 2 subunit control calpain-mediated cleavage of the NMDA receptor. J Neurosci. 2004; 24(49): 11035-45.
- Fenner BM. Truncated TrkB: beyond a dominant negative receptor. Cytokine Growth Factor Rev. 2012; 23(1-2):15-24.
- Fischer A, Sananbenesi F, Pang PT, Lu B, Tsai LH. Opposing roles of transient and prolonged expression of p25 in synaptic plasticity and hippocampus-dependent memory. Neuron. 2005; 48(5):825-38
- Gafni J, Ellerby LM. Calpain activation in Huntington’s disease. J Neurosci. 2002; 22: 4842–4849.
- Gladding CM, Sepers MD, Xu J, et al. Calpain and STriatal-Enriched protein tyrosine phosphatase (STEP) activation contribute to extrasynaptic NMDA receptor localization in a Huntington's disease mouse model. Hum Mol Genet. 2012;21(17):3739-52.
- Goll DE, Thompson VF, Li H, Wei W. The calpain system. J Physiol Rev. 2003; 83 (3): 731–801.
- Gomes JR, Costa JT, Melo CV, et al. Excitotoxicity downregulates TrkB.FL signaling and upregulates the neuroprotective truncated TrkB receptors in cultured hippocampal and striatal neurons. J Neurosci. 2012;32(13):4610-22.
- Hackam AS, Singaraja R, Wellington CL, et al. The influence of huntingtin protein size on nuclear localization and cellular toxicity. J Cell Biol. 1998;141(5):1097-105.
- Hardingham, G.E., Bading, H., (2010). Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat. Rev. Neurosci. 11 (10), 682–696.
- Hu Y, Russek SJ. BDNF and the diseased nervous system: a delicate balance between adaptive and pathological processes of gene regulation. J Neurochem. 2008;105(1):1-17.
- Huang EJ, Reichardt LF. Trk receptors: roles in neuronal signal transduction. Annu Rev Biochem. 2003;72:609-42.
- Jerónimo-Santos A, Vaz SH, Parreira S, Rapaz-Lérias S, Caetano AP, Buée-Scherrer V, et al. Dysregulation of TrkB receptors and BDNF function by amyloid-β peptide is mediated by calpain. Cereb Cortex. 2015; 25 (9), 3107–3121.
- Kalb R. The protean actions of neurotrophins and their receptors on the life and death of neurons. Trends In Neurosciences. 2005; 28 (1): 5-11.
- Kanning KC, Hudson M, Amieux PS, Wiley JC, Bothwell M, Schecterson LC. Proteolytic processing of the p75 neurotrophin receptor and two homologs generates C-terminal fragments with signaling capability. J Neurosci. 2003;23(13):5425-36.
- Kaplan DR, Miller FD. Neurotrophin signal transduction in the nervous system. Curr Opin Neurobiol. 2000;10(3):381-91.
- Klein R, Conway D, Parada LF, Barbacid M. The trkB tyrosine protein kinase gene codes for a second neurogenic receptor that lacks the catalytic kinase domain. Cell. 1990;61(4):647-56.
- Klein R, Nanduri V, Jing SA, et al. The trkB tyrosine protein kinase is a receptor for brain-derived neurotrophic factor and neurotrophin-3. Cell. 1991;66(2):395-403.
- Lew J. CDK5: A new lead to survival. Cell Cycle. 2013; 12(13): 1981–1982.
- Liu Y, Dore J, Chen X. Calcium influx through L-type channels generates protein kinase M to induce burst firing of dopamine cells in the rat ventral tegmental area. J Biol Chem. 2007;282(12):8594-603.
- Liu J, Liu MC, Wang KK. Calpain in the CNS: from synaptic function to neurotoxicity. Sci Signal. 2008;1(14):re1.
- Lu X, Wyszynski M, Sheng M, Baudry M. Proteolysis of glutamate receptor-interacting protein by calpain in rat brain: implications for synaptic plasticity. J Neurochem. 2001;77(6):1553-60.
- Lu B, Pang P, Woo, N. The Yin And Yang Of Neurotrophin Action. Nature Reviews Neuroscience. 2005; 6: 603-614.
- Ma M. Role of calpains in the injury-induced dysfunction and degeneration of the mammalian axon. Neurobiol. Dis. 2013; 60, 61–79.
- Masoudi R, Ioannou MS, Coughlin MD, et al. Biological activity of nerve growth factor precursor is dependent upon relative levels of its receptors. J Biol Chem. 2009;284(27):18424-33.
- Mcnamara JO, Huang YZ, Leonard AS. Molecular signaling mechanisms underlying epileptogenesis. Sci STKE. 2006;2006(356):re12.
- Neukomm LJ, Freeman MR. Diverse cellular and molecular modes of axon degeneration. Trends Cell Biol. 2014;24(9):515-23.
- Patrick GN, Zukerberg L, Nikolic M, De la monte S, Dikkes P, Tsai LH. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature. 1999;402(6762):615-22.
- Tejeda GS, Ayuso-Dolado S, Arbeteta R, Esteban-Ortega GM, Vidaurre OG, Díaz-Guerra M. Brain ischaemia induces shedding of a BDNF-scavenger ectodomain from TrkB receptors by excitotoxicity activation of metalloproteinases and γ-secretases. J Pathol. 2016; 238(5):627-40.
- Tymianski M, Charlton MP, Carlen PL, Tator CH. Source specificity of early calcium neurotoxicity in cultured embryonic spinal neurons. J Neurosci. 1993;13(5):2085-104.
- Unsain N, Montroull LE, Mascó DH. Brain-derived neurotrophic factor facilitates TrkB down-regulation and neuronal injury after status epilepticus in the rat hippocampus. J Neurochem. 2009;111(2):428-40.
- Unsain N, Nuñez N, Anastasía A, Mascó DH. Status epilepticus induces a TrkB to p75 neurotrophin receptor switch and increases brain-derived neurotrophic factor interaction with p75 neurotrophin receptor: an initial event in neuronal injury induction. Neuroscience. 2008;154(3):978-93.
- Vidaurre OG, Gascón S, Deogracias R, et al. Imbalance of neurotrophin receptor isoforms TrkB-FL/TrkB-T1 induces neuronal death in excitotoxicity. Cell Death Dis. 2012;3:e256.
- Vinade L, Petersen JD, Do K, Dosemeci A, Reese TS. Activation of calpain may alter the postsynaptic density structure and modulate anchoring of NMDA receptors. Synapse. 2001;40(4):302-9.
- Xie W, Song YJ, Li D, Pan LP, Wu QJ, Tian X. The suppression of epileptiform discharges in cultured hippocampal neurons is regulated via alterations in full-length tropomyosin-related kinase type B receptors signaling activity. Eur J Neurosci. 2014; 40 (3): 2564–2575.
- Xu W, Wong TP, Chery N, Gaertner T, Wang YT, Baudry M. (2007) Calpain-mediated mGluR1alpha truncation: a key step in excitotoxicity. Neuron. 53(3):399-412.
- Yildiz-Unal A, Korulu S, Karabay A. Neuroprotective strategies against calpain mediated neurodegeneration. Neuropsychiatr Dis Treat. 2015; 11, 297–310.