
Recent advances in multiple system atrophy
Kurt A. Jellinger
Abstract
Multiple system atrophy (MSA) is a fatal orphan neurodegenerative disorder that manifests with autonomic, parkinsonian, cerebellar, and pyramidal features. It is characterized by the accumulation of misfolded α-synuclein (αSyn) in oligodendroglia and neurons, affecting multiple parts of the central, autonomic and perípheral system. Both the etiology and pathogenesis of MSA are unknown, although a genetic component has been proposed. Accumulation of aberrant αSyn in oligodendrocytes, preceded by relocation of p25α protein from myelin into oligodendroglia, results in the formation of insoluble glial cytoplasmic inclusions (GCIs). These changes are associated with proteasomal, mitochondrial and lipid transport dysfunction, oxidative stress, reduced trophic transport, neuroinflammation and other noxious factors. Their interaction induces dysfunction of the oligodendroglial-myelin-axon-neuron complex, resulting in the system-specific pattern of neurodegeneration characterizing MSA as a synucleinopathy with oligodendroglio-neuronopathy. Propagation of modified toxic αSyn species from neurons to oligodendroglia by "prion-like" transfer and its spreading to associate neuronal pathways result in multi-system involvement. No reliable biomarkers are currently available for the clinical diagnosis and prognosis of MSA and neither effective neuroprotective nor disease-modifying therapies of MSA are available, although novel treatment strategies targeting αSyn are under discussion. Multidisciplinary research to elucidate the genetic and molecular background of the deleterious cycle of noxious processes to develop reliable diagnostic biomarkers and to deliver targets for effective treatment of this hitherto incurable disorder is urgently needed.
Introduction
Multiple system atrophy (MSA) is a rare, adult-onset, progressive, fatal neurodegenerative disorder of uncertain etiology that presents clinically with a variable combination of autonomic, parkinsonian, cerebellar and pyramidal features1. Together with Parkinson disease (PD) and Lewy body dementia (DLB), MSA belongs to the neurodegenerative group of α-synucleinopathies, which are characterized by accumulation of misfolded α-synuclein(αSyn)2,3. MSA is subdivided into (1.) a parkinsonian variant (MSA-P) associated with striatonigral degeneration (SND) (2.) the cerebellar subtype with olivopontocerebellar atrophy (OPCA) if cerebellar features predominate4 and (3.) a combination of the two, referred to as mixed MSA5. The annual incidence in the 50-99 years age group is 3 cases/100,000 persons; the estimated point prevalence is 1.9 to 4.9 cases, increasing to 7.8/100,000 persons per year over age 50 (PD is about 45 times more common). In the Western hemisphere, MSA-P involves about 70%, whereas in Asian populations, the cerebellar subtype (MSA-C) predominates in two-thirds of the patients, with genetic or epigenetic factors possibly exerting an influence6. The different symptom assessment scales for MSA have been compared7. Motor symptom onset is 56±9 years, however, non-motor features including cardiovascular autonomic failure, respiratory and urogenital disorders may precede the motor symptoms by some years7,8. The prevalence of rapid eye movement (REM) sleep disorders in MSA is up to 88%9. The mean survival from symptom onset of 6 to 10 (mean 9.5) years10 is similar for both phenotypes11, while in Japan, MSA-C had a better prognosis than patients with Parkinsonism or autonomic failure at onset12.
Recent consensus criteria differentiate possible, probable and definite MSA, the latter confirmed by postmortem examination4,13. Two or more of six red flags (warning signs) had a specificity of 98.3% and a sensitivity of 84.2%14. Several subtypes, e.g., "minimal-change" aggressive and prolonged "benign" forms do not fit into the current classification11,15-19. Due to overlapping clinical presentation, the distinction between MSA, early disease PD or atypical parkinsonian disorders (DLB, progressive supranuclear palsy/PSP) may be difficult20-22. A recently described atypical MSA (aMSA) was identified as frontotemporal lobar degeneration with αSyn (FTLD-synuclein)23,24.
Etiology
The causes of MSA are unknown. Epidemiological studies are limited by low case numbers and diagnostic difficulties25,26. MSA is generally a sporadic disorder, but there are familial cases, and in some pedigrees, the disease has been transmitted in an autosomal dominant or recessive inheritance pattern27,28. A recent genome-wide estimate suggested MSA heritability due to common risk variants at 2.9 to 6.65%29. No definite risk factors have been identified, although mutations of the COQ2 gene have been linked to MSA in Japanese families30-32, and SNCA (encoding αSyn and other loci) have been associated with increased risk for MSA33,34. However, these findings were not replicated by others29,35,36. The presence of GCI-like oligodendroglial inclusions in familial PD due to SNCA mutations also suggests that PD and MSA form a continuum of αSyn pathology37. Gaucher disease causing glucocerebrosidase gene (GBA) variants38 but neither association with GBA mutations39 nor with C9orf72 hexanucleotide repeat expansions have been found40, whereas contribution of LRRK2 exonic variants to susceptibility of MSA are under discussion41,42. RNA sequencing analyses of MSA brain have revealed alterations in a number of genes including α and β hemoglobin43 and uncovered disruption of line RNAs along with protein coding genes related to iron metabolism and immunological response regulation, indicating complexity in transcriptional pathology of MSA44. A widespread dysregulation of microRNAs resulted in downregulation of the carrier family SLC1A1 and SLC645. RNA analysis of MSA brain uncovered a wide dysregulation of microRNAs (mRNA) resulting in downregulation of the carrier protein family SLC1A1 and SLC6A645. The role of mRNA in the pathogenesis of neurodegenerative disorders has been summarized recently46. Genome-wide changes of αSyn mRNA expression may be important for the increased deposition of αSyn in MSA brain31,47, but the lack of causative mutations makes generating valid models of MSA difficult48. G51D SNCA and A53E single point mutations, as well as SNCA triplications, give rise to a histopathological phenotype with inclusions in both neuronal and oligodendroglial cells comparable to both PD and MSA49-51. Sparse evidence supports the notion that epigenetic factors or environmental toxins may play a role in MSA development26,52, which was not confirmed in a tg MSA mouse model53.
Neuropathology
The histological core features of MSA are (1) four types of cellular αSynimmuno reactive inclusions, i.e. GCI within oligodendrocytes (the presence of which is required for the postmorten diagnosis of definite MSA13, less frequent glial nuclear (GNI), neuronal cytoplasmic (NCI), and neuronal nuclear inclusions (NNI), (2) astroglial cytoplasmic inclusions and neuronal threads, also composed of αSyn55, (3) selective neuronal loss and axonal degeneration involving multiple regions of the nervous sytem54, and (4) myelin degeneration with reduction of myelin basic protein (MBP), both with accompanying astrogliosis55. GCIs and the resulting neurodegeneration occur in typical multisystemic distribution encompassing the striatonigral and OPC systems, autonomic nuclei of the brainstem (locus ceruleus, nucleus raphe, dorsal vagal nuclei, etc.), spinal cord and sacral visceral sensory pathways56-59.
The biochemical composition and distribution of GCIs and other inclusions have been summarized recently2,60-62. Based on semiquantitative assessment of GCIs and neuronal loss, the striatonigral and OPC lesions were assessed into four degrees of severity63, but there is an overlap between the degeneration of both systems64. The number of GCIs increases with disease duration, and there is a positive correlation between their density and the degree of neurodegeneration60,65. Region-specific astrogliosis is positively correlated with αSyn pathology in MSA in contrast to PD66. Glial and neuronal inclusions also involve many other parts of the central and peripheral nervous system underpinning the multisystem character of MSA2,36,37. The thickness of the retinal fiber and macular ganglion cell complex is reduced68. Accumulation of phosphorylated αSyn also occurs in subpial and periventricular astrocytes after long disease duration69,70. Cortical and subcortical gray matter atrophy in MSA-P and neocortical neuronal loss may underly cognitive impairment in MSA71,75, which may also associated with the presence of Lewy-body like inclusions in neocortex67, while others found no pathological changes related to cognitive impairment in MSA76.
Demyelination in MSA mainly involves the striatonigral and OPC regions and is associated with reduction of myelin proteins77, but no portion of the nervous system appears to be spared. Whether myelin loss is secondary to neuronal or axonal loss or whether it is a primary lesion, which in turn leads to neuronal and axonal loss, is unknown. White matter degeneration causes a destruction of neuronal loops, resulting in dysfunction of the whole-brain network78, and may be related to disorders of cerebral autoregulation79. Differences in expression of a disease-related metabolic pattern correlate with disease severity in MSA80.
αSyn pathology has also been detected in Schwann cells of cranial, spinal and autonomic nerves70,81, inducing postganglionic sudomotor denervation82, and in neuronal cytoplasm and processes of sympathetic ganglia (in 42.3% of MSA cases)83, causing multidimensional autonomic failure84. In the peripheral nervous system, αSyn aggregates have been observed in sympathetic ganglia, skin nerve fibers85,86 and Schwann cells70, which have also been described in MSA models53, whereas other studies showed absence of phosphorylated αSyn immunoreactivity in dermal nerve fibers in contrast to PD85,87.
α-Synuclein in MSA
αSyn, a heat stable cytosolic protein, primarily located in presynaptic nerve terminals, when present in oligodendrocytes in MSA and tg models, has undergone post-translational modifications (oxidation, nitration, phosphorylation, etc.) enhanced by oxidative stress (OS)88-90. αSyn in GCIs of MSA is phosphorylated at residue Ser-129 and ubiquinated as is the case in Lewy bodies (LBs)91,92. Elevated levels of membrane-associated detergent-soluble αSyn were seen in disease-affected regions of MSA brains containing both neuronal and glial inclusions 5- to 10-fold higher than in patients with PD93. However, most of the soluble αSyn was also present in areas with few GCIs, suggesting that altered solubility precedes the formation of GCIs and that increased soluble monomeric αSyn may result in a conversion into insoluble, filamentous αSyn aggregation, which could result in neurodegeneration94. MSA brain contains various levels of αSyn isoforms, 140 and 112 isoforms being significantly increased, whereas αSyn 126 isoform was decreased95,96. This corresponds to the regional pathology, including GCI distribution, although accumulation of monomeric αSyn may not be merely caused by the formation of GCIs. MSA brain also containes increased levels of parkin isoforms and an aggregation-prone synphilin-1A isoform. This suggests alterations in protein-protein interactions that may be important in aggregation processes and may result in neurotoxicity and/or neuroinflammation95. Autophagy is involved in the protein aggregate formation in MSA oligodendroglia97. There is widespread mRNA dysregulation in MSA patients, which is recapitulated in murine models45, and circulating mRNAs discriminate MSA from PD98. Inhibition of UCH-L1 in oligodendrocytes resulting in microtubule stabilization may prevent αSyn aggregate formation by activating the autophagic pathway99.
Accumulation of αSyn in oligodendrocytes may induce their dysfunction resulting in reduced trophic support and demyelination, as suggested by the MBP-hαSyntg mouse model100,101. Most affected regions are basal ganglia and cerebellum, with loss of myelin staining, astrogliosis, and axonal alterations, correlating with the severity of both the GCI pathology and the level of αSyn expression. The leading role of GCI pathology is supported by the "minimal change" (MC-MSA) forms, where severe GCI burden is associated with less severe neuronal loss but shorter disease duration18.
Changes in MBP levels in MSA brains suggest myelin lipid dysfunction and instability77,101,102, which, together with an aberration in protein distribution may lead to myelin dysfunction in MSA103. Region-specific increased matrix metalloproteinase activity may contribute to the disease progress by promoting blood-brain barrier dysfunction and myelin degradation104. Oligodendrocytes differentiated from MSA-derived stem cells have been suggested to express αSyn in contrast to those derived from healthy controls or PD patients, but suppress its expression during maturation105,106, indicating the possibility that an endogenous intra-oligodendroglial αSyn source may contribute to the GCI formation and thus they may play a primary role in triggering neurodegeneration. Oligodendroglial precursor cells (OPCs) show increased density in MSA white matter affected by GCI pathology and myelin degeneration suggesting repair (remyelination) efforts107. While αSyn has been shown to impair oligodendrocyte progenitor maturation in MSA preventing the formation of mature oligodendroglial cells108,109, others have shown a link between the intracellular level of human αSyn and the maturation of primary OPCs108. The data on oligodendroglial dysfunction in MSA support the notion that neurodegeneration may occur secondary to demyelination and lack of trophic support by GCI-bearing oligodendroglia.
Transgenic animal models of MSA
The causative role of GCI-like pathology for the induction of neurodegeneration in MSA was confirmed experimentally in tg mice expressing human αSyn in oligodendrocytes under various oligodendroglia-promotors90,100,110,111. The selectivity of glial and neuronal degeneration in human MSA and in these models is still unresolved, since there are regional disparities between mouse lines. While one model demonstrated most severe pathology in the spinal cord, PLP-αSyntg mice90 showed the classical distribution with basal ganglia, cerebellum and autonomic centers severely affected110-113. The regional disparities between mouse lines sharing the same basic defect could result from different efficiencies of the promoter expressing αSyn or because the various promoters target different subtypes of oligodendrocytes. Since none of the available tg mouse lines reproduced the specific predominance of SND or OPCA in human disease62, a more accurate characterization of the mouse models of MSA is required.
Prion-like transmission of αSyn
The source of αSyn in GCIs in oligodendrocytes and the role of many of their polypeptides and protein components are enigmatic, although these have been shown to express αSyn mRNA105. αSyn, previously suggested to be an exclusively neuronal protein, can be transferred to grafted oligodendrocytes from host rat brain neurons overexpressing αSyn, supporting a neuron-to-oligodendrocyte transfer114. Three possible causes were discussed – redistribution of αSyn from neurons to oligodendrocytes, suppression of neuronal expression from oligodendrocytes, and neuronal clearance of αSyn. αSyn released from degenerating neurons both mediates formation of abnormal inclusion bodies and induces neuroinflammation, which might also favor the formation of intracellular αSyn aggregates as a consequence of cytokine release and the shift to a pro-inflammatory environment115. Pathogenic mechanisms leading to elevated αSyn in neurons underly neuronal secretion and subsequent uptake of αSyn by oligodendroglia in MSA116.
Recent evidence suggests that – similar to preclinical models of PD – αSyn spreads through the brain in a "prion-like" manner in MSA to other functionally connected neuronal networks117, resulting in a system-like pattern of neurodegeneration that is typical of MSA118. "Prion-like" seeding, introduction of toxic environment, and intrinsic disruption of proteostasis may synergetically contribute to the induction and spread of αSyn inclusions119. MSA may be caused by a unique strain of αSyn proteins, which is different from the putative prions causing PD and from those causing spontaneous neurodegeneration in TgM83(+/+) mice120. However, the same studies failed in wildtype/healthy mouse brain and no oligodendroglial αSyn aggreation was seen, therefore the core pathology of MSA could not be reproduced. Experimental studies showed that oligodendrocytes may take up αSyn from the extracellular space114, but no typical GCI-like aggregations were observed. Primary oligodendroglial dysfunction may therefore result in ectopic accumulation of αSyn in these cells60. Alternatively, specific αSyn conformational strains were proposed to be responsible for the degeneration of MSA-like αSyn seeding121. However, this needs further verification, as inoculation of MSA-derived αSyn into brains of healthy, non-tg mice did not induce GCI-like pathology120, and none of the available models has so far achieved the phenotypic resemblance to true MSA122. Hence, aside from α-SYN strains that can expose distinct interaction surfaces and spread between cells, additional factors are required in order to drive a complete MSA phenotype. These processes are likely multifactorial and potentially driven by the presence of αSyn strains on a background that promotes but also sustains the formation of GCIs123.
Pathogenic mechanisms of MSA
Evidence from animal models and human postmortem studies suggest that the accumulation of misfolded αSyn plays a central role in the disease process58,62, which can be considered a synucleinopathy with specific glioneuronal degeneration, associated with early myelin dysfunction and neuronal degeneration related to retrograde axonal disease55,124. Although one may speculate that primary neuronal pathology leads to secondary oligodendroglia degeneration as suggested by the finding that NCIs exist in areas lacking GCIs67, the fact that distribution and severity of neurodegeneration reflect sub-regional GCI densities supports the hypothesis of a primary oligodendrogliopathy60,62. In early stages of the disease, in addition to GCIs, diffuse homogenous αSyn staining in neuronal nuclei and cytoplasm was observed in many parts of the central nervous system. The density of the GCIs is unrelated to that of NNIs125. The number of NNIs was much larger than the NCI count in the pontine nuclei in some MSA cases, suggesting that NCI formation is accelerated by the progression of the disease process and that NNI formation may be an earlier phenomenon than NCI formation15.
The earliest stages of MSA pathogenesis are likely to involve a relocation of p25α (TPPP - tubulin polymerization-promoting protein), an oligodendroglia-specific phosphoprotein and stabilizer of microtubules and myelin integrity126, from the myelin sheaths to the oligodendroglial soma. This is associated with myelin dysfunction, reduction of full-length MBP, demyelination of small-caliber axons and an increase in oligodendroglial soma size, preceding αSyn aggregation127. The interaction between p25α and αSyn promotes phosphorylation and aggregation into insoluble oligomers and GCIs implies that mitochondrial dysfunction can lead to secondary p25α relocation, probably linked to dysregulation of lipid metabolism and dysfunctional myelination, probably a fundamental event in MSA pathogenesis128.
The aggregation of αSyn is suggested to interfere with the process of oligodendrogenesis, preventing the formation of mature oligodendroglia cells108,109. Transgenic oligodendroglial expression of human αSyn causing a phenotype that resembles human MSA demonstrated that accumulation of αSyn in oligodendroglia induces subsequent degeneration of both oligodendroglia and neurons100,129. Enhanced FAS (Fas cell surface death receptor) gene expression is an early hallmark of oligodendroglial pathology in MSA that may be related to αSyn-dependent degeneration130,131. Predegenerative expression of the transcription factor I?Bα as an early event in the course of MSA may cause destructive neuroinflammation tissue responses, and thus contribute to cellular demise130. Recently, differential involvement of the cysteine protease inhibitor cystatin C, associated with increased risk for neurodegeneration, has been observed in MSA phenotypes, suggesting its role in the pathogenesis of this disorder132.
Formation of GCIs interferes with oligodendroglial and neuronal trophic support leading to death of these cells and to initiation of neuroinflammation by activation of quiescent microglia133, suggesting that pathological αSyn triggers neuroinflammatory responses in the MSA brain60,134. Microglial activation may contribute to the progression of the neurodegenerative process in MSA via increased levels of reactive oxygen species in degenerating areas135. Currently available data support the hypothesis that misfolded αSyn contributes to OS through a pathway that induces microglia activation as well as antioxidant responses and requires an additional protein structure136, but OS appears unlikely to be the sole mechanism for αSyn aggregation.
The cell death mechanisms in MSA are poorly understood. Increased iron levels in degenerating brain areas suggest that OS may play a significant role in the selected neuronal loss in MSA, and microglial activation may contribute to the increased levels of reactive oxygen species in the degenerating areas135. Loss of phosphoprotein DARPP-32 and calbindin-D 28k in areas of less prominent / absent neuronal loss indicates calcium toxicity and disturbance of the phosphorylated state of proteins as relatively early events137. While experimental studies support the dysfunction of the proteasome and autophagosome systems in oligodendroglial α-synucleinopathy97,138, excitotoxic cell death was not aggravated by GCI pathology53.
Various mechanisms possibly related to cell death include X-linked inhibitor of apoptosis protein (XIAP) that is upregulated in GCI- and NCI-bearing oligodendrocytes and neurons139, proteasomal140 or autophagosomal dysfunction141, supported by experimental studies97,101,135. Neuronal death may also be related to an altered communication between neurons and oligodendrocytes due to perturbation of their neurotrophic transport142.
The burden of neuronal pathology appears to increase multifocally as an effect of disease duration associated with increasing overall αSyn burden. A correlation between neuronal pathology and GCIs and NIs in the severely affected brain regions suggests a link beween these phenomena5, although the underlying mechanisms remain to be elucidated.
In conclusion, although the pathogenesis of MSA is currently poorly understood, evidence from animal models and human postmortem studies suggest that the accumulation of misfolded αSyn plays a central role in the disease process58,62, which can be considered a synucleinopathy with specific glioneuronal degeneration, associated with early myelin dysfunction and neuronal degeneration related to retrograde axonal disease55,124 (Figure 1). Although one may speculate that primary neuronal pathology leads to secondary oligodendroglia degeneration as suggested by the finding that NCIs exist in areas lacking GCIs67, the fact that distribution and severity of neurodegeneration reflect subregional GCI densities supports the hypothesis of a primary oligodendrogliopathy60,62.
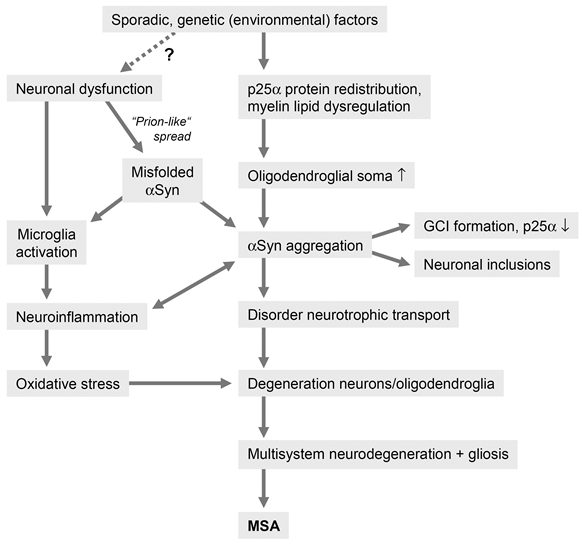
Figure 1: Putative pathogenic pathways of MSA
MSA has also been suggested to be a primary neuronal disease, the formation of GCIs probably resulting from secondary accumulation of pathological αSyn that may be neuronal in origin142. However, strong evidence against a primary neuronal pathology is the fact that GCIs are the hallmark of MSA and not in PD, a disease with similar lesion pattern of αSyn immuno reactive neuronal inclusions in many overlapping circuits but few GCIs, which differentiates these two disorders133. Although the source of αSyn is under discussion, "prion-like" spreading of this protein, OS, proteasomal and mitochondrial dysfunction45 and proteolyticdys balance, dysregulation of myelin lipids, demyelination, neuroinflammation and energy failure and genetic polymorphism47 are suggested to contribute to the pathogenesis of systemic neurodegeneration in this unique proteinopathy143,144.
Potential biomarkers of MSA
Recent international consensus criteria allow the diagnosis of MSA with three levels of certainty: a diagnosis of possible and probable MSA is based on the presence of clinical core features, while that of definite MSA requires postmortem confirmation4. In a series of neurodegenerative diseases, the presence of neuroimaging and/or fluid (serum, cerebrospinal fluid/CSF) biomarkers has improved the diagnostic accuracy, prognostic guidance and may serve as efficacy measures or surrogates of target engagement for clinical trials145-152. Biomarkers include those based on neuroimaging, in particular multimodal magnetic resonance imaging (MRI), diffusion tensor imaging (DTI), MR spectroscopy, and positron emission tomography (PET), peripheral biomarkers (skin, peripheral nerve and other biopsies) and fluid biomarkers from serum, plasma and CSF. In general, there is no single biomarker that will satisfy all requirements, whereas combinations are likely to be of great value in improving the diagnostic accuracy and prognostic guidance146. Unfortunately, despite growing research efforts, no biomarker currently exists for the diagnosis and prognosis of MSA.
Modern neuroimaging techniques have improved clinical accuracy in differentiating MSA and PD. MRI abnormalities in PD are subtle, whereas MSA shows more obvious imaging abnormalities such as the "hot cross bun" (HBC) sign (selective loss of myelinated transverse pontocerebellar fibers in the pontine raphe with selective preservation of the corticospinal tracts), atrophy of the pons, cerebellum, and globuspallidus153,154, although no single feature is completely sensitive and specific. The same holds for MR spectroscopy and PET155. Multimodal MRI reveals different patterns of nigro-striatal involvement between PD and MSA156, while others found significant differences of FA/RD values in bilateral corticospinal tract (CST) and left anterior thalamic radiation (ATR) in MSA-P versus PD and controls157,158. Hypointensity of the dorsolateral putamen in T2-weighted MRI due to iron deposition and more severe white matter abnormalities differentiate MSA-P from MSA-C75, while dopamine transporter single-photon emission computerized tomography (DAT-SPECT) cannot differentiate MSA from PD158. Automated subcortical volume measurement can be used for differentiating MSA from PD and PSP with high accuracy160. Since sympathetic denervation in PD involves postganglionic neurons, while in MSA affects preganglionic neurons, cardiac 123Imetaiodobenzylguanidine (MIBG) scintigraphy may also be useful for the differential diagnosis of these disorders161, but this test has suboptimal diagnostic accuracy1.
The diagnostic validity of skin punch biopsies for the demonstration of αSyn deposits in Schwann or other cells in peripheral nerves in MSA patients is under discussion and needs further evaluation in pathologically confirmed cases69,85,86.
The MSA Biomarker Initiative has recently published a critical review of fluid markers based on the evaluation of 60 studies162, suggesting that combining several CSF fluid biomarkers may be more successful than using single markers. Currently, clinically most useful is a combination of the light chain of neurofilaments, which is consistently elevated in MSA compared to controls and PD163, metabolites of the catecholamine pathway (dopamine and norepinephrine) and proteins such as αSyn, DJ-1, and total-tau (t-tau). A panel associating DJ-1 + t-tau + phosphorylated tau (p-tau) yielded high sensitivity (82%) and specificity (91%) for the distinction between MSA and PD or controls164,165. The best performance was achieved by combining NFL and FLT3 ligand163, while the combination of DJ-1 + t-tau + p-tau protein in CSF improved the discrimination between MSA from PD and controls164,166. The results of proteomics for biomarker discovery and of miRNA expression need validation using independent technologies162.
Only a few studies revealed a relation between fluid biomarker levels and clinical features, while most did not collect detailed clinical data to allow such and almost no studies had postmortem confirmation. Further research to elucidate the molecular background of the development and progression of the disease process of MSA are urgently needed in order to unmask the interplay of the various pathobiological changes as a basis for the development of reliable biomarkers and as an emerging template for the development of disease-modifying treatment options of this hitherto incurable disorder.
Modern treatment strategies in MSA
Currently, there is neither an effective neuroprotective nor a disease-modifying therapy in MSA. Although several pharmacological approaches have been tried in transgenic mouse or cellular models of MSA, including riluzole, rasagiline, minocycline, rifampicin, stem cells, etc., treatments that can halt or reverse the disease progression in humans have not yet been identified167-170. Symptomatic approaches include dopaminergic and anticholinergic agents, amentadine, paroxetine, non-pharmacological treatment, treatment of orthostatic hypotension, urinary and erectile dysfunction as well as palliative care171,172. Active immunization against αSyn has been shown to ameliorate the degenerative pathology and to prevent demyelination in a mouse model of MSA173, while a modified brain-targeted neurosin (kallikrein-6) that reduces αSyn accumulation in an MSA mouse model may warrant further investigations as potential therapy for MSA174, postmortem assessment of the short- and long-term effects of gene delivery of the trophic factor neurturin in patients with α-synucleinopathies (including one MSA-P case) showed its mild but persistent expression over 4 years175. Prominent targets for disease therapy include (1) pathological αSyn accumulation, (2) microglia activation and neuroinflammation, (3) oligodendroglial dysfunction, and (4) cell death58. Combination therapies, eg, immunotherapy against αSyn + antiinflammatory agents or multi-target drugs may be the next step for the treatment of synucleinopathies176. A new European project (SYMPATH) is currently assessing a vaccine targetting αSyn (AFFITOPE) in PD and MSA in humans168.
Deep brain stimulation could not be recommended for MSA patients177. Emerging targets for interventional therapies of MSA were summarized recently58, and therapeutic strategies targeting αSyn in vivo and in vitro models of MSA (αSyn expression, aggregation, degradation and clearance as well as cell-to-cell propagation) as possible basis for effective disease-modifying alternatives were critically reviewed168,178.
Further research on the pathogenic mechanisms, the interplay of the disease process with various molecular changes, and the nature of possible genetic and environmental triggers that unmask its pathogenesis will be needed to develop optimal animal models, and to clarify the relations between the development of pathomorphology and clinical manifestations as a basis for early diagnosis and a disease-modifying treatment of this hitherto incurable devastating disorder.
Acknowledgements
The study was supported in part by the Society for the Promotion of Research in Experimental Neurology, Vienna, Austria. The author thanks Mr. E. Mitter-Ferstl, PhD, for secretarial and computer work.
References
- Fanciulli A, Wenning GK. Multiple-system atrophy. N Engl J Med. 2015; 372:249-263.
- Jellinger KA. Neuropathology of multiple system atrophy: New thoughts about pathogenesis. Mov Disord. 2014; 29:1720-1741.
- Spillantini MG, Goedert M. Synucleinopathies: past, present and future. Neuropathol Appl Neurobiol. 2016.
- Gilman S, Wenning GK, Low PA, Brooks DJ, Mathias CJ, Trojanowski JQ, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology. 2008; 71:670-676.
- Ozawa T, Paviour D, QuinnNP, Josephs KA, Sangha H, Kilford L, et al. The spectrum of pathological involvement of the striatonigral and olivopontocerebellar systems in multiple system atrophy: clinicopathological correlations. Brain. 2004; 127:2657-2671.
- Matsushima M, Yabe I, Oba K, Sakushima K, Mito Y, Takei A, et al. Comparison of different symptom assessment scales for multiple system atrophy. Cerebellum. 2016; 15:190-200.
- Jecmenica-Lukic M, Poewe W, Tolosa E, Wenning GK. Premotor signs and symptoms of multiple system atrophy. Lancet Neurol. 2012; 11:361-368.
- Xie T, Kang UJ, Kuo S-H, Poulopoulos M, Greene P, Fahn S. Comparison of clinical features in pathologically confirmed PSP and MSA patients followed at a tertiary center. npj Parkinson's Dis. 2015; 1.
- Palma JA, Fernandez-Cordon C, Coon EA, Low PA, Miglis MG, Jaradeh S, et al. Prevalence of REM sleep behavior disorder in multiple system atrophy: a multicenter study and meta-analysis. Clin Auton Res. 2015; 25:69-75.
- Petrovic IN, Ling H, Asi Y, Ahmed Z, Kukkle PL, et al. Multiple system atrophy-parkinsonism with slow progression and prolonged survival: a diagnostic catch. Mov Disord. 2012; 27:1186-1190.
- Roncevic D, Palma JA, Martinez J, Goulding N, Norcliffe-KaufmannL, Kaufmann H. Cerebellar and parkinsonian phenotypes in multiple system atrophy: similarities, differences and survival. J Neural Transm (Vienna). 2014; 121:507-512.
- Sakushima K, Nishimoto N, Nojima M, Matsushima M, Yabe I, Sato N, et al. Epidemiology of multiple system atrophy in Hokkaido, the northernmost island of Japan. Cerebellum. 2015; 14:682-687.
- Trojanowski JQ, Revesz T. Proposed neuropathological criteria for the post mortem diagnosis of multiple system atrophy. Neuropathol Appl Neurobiol. 2007; 33:615-620.
- Köllensperger M, Geser F, Ndayisaba JP, Boesch S, Seppi K, Ostergaard K, et al. Presentation, diagnosis, and management of multiple system atrophy in Europe: final analysis of the European multiple system atrophy registry. Mov Disord. 2010; 25:2604-2612.
- Wakabayashi K, Mori F, Nishie M, Oyama Y, Kurihara A, Yoshimoto M, et al. An autopsy case of early ("minimal change") olivopontocerebellar atrophy (multiple system atrophy-cerebellar). Acta Neuropathol. 2005; 110:185-190.
- Kon T, Mori F, Tanji K, Miki Y, Wakabayashi K. An autopsy case of preclinical multiple system atrophy (MSA-C). Neuropathology. 2013; 33:667-672.
- Masui K, Nakata Y, Fujii N, Iwaki T. Extensive distribution of glial cytoplasmic inclusions in an autopsied case of multiple system atrophy with a prolonged 18-year clinical course. Neuropathology. 2012; 32:69-76.
- Ling H, Asi YT, Petrovic IN, Ahmed Z, Prashanth LK, Hazrati LN. Minimal change multiple system atrophy: An aggressive variant? Mov Disord. 2015; 30:960-967.
- McCann H, McGeachie AB, Silberstein P, Lewis SJ, Halliday GM. Restricted disease propagation in multiple system atrophy with prolonged survival. Neuropathol Appl Neurobiol. 2015; 41: 681-685.
- Kim HJ, Jeon BS, Jellinger KA. Diagnosis and differential diagnosis of MSA: boundary issues. J Neurol. 2015a; 262:1801-1813.
- Kim HJ, Stamelou M, Jeon B. Multiple system atrophy-mimicking conditions: Diagnostic challenges. Parkinsonism Relat Disord. 2016; 22 S12-15.
- Koga S, Aoki N, Uitti RJ, van Gerpen JA, Cheshire WP, Josephs KA, et al. When DLB, PD, and PSP masquerade as MSA: an autopsy study of 134 patients. Neurology. 2015; 85: 404-412.
- Aoki N, Boyer PJ, Lund C, Lin WL, Koga S, Ross OA, et al. Atypical multiple system atrophy is a new subtype of frontotemporal lobar degeneration: frontotemporal lobar degeneration associated with alpha-synuclein. Acta Neuropathol. 2015; 130:93-105.
- Rohan Z, Rahimi J, Weis S, Kapas I, Auff E, Mitrovic N, et al. Screening for alpha-synuclein immunoreactive neuronal inclusions in the hippocampus allows identification of atypical MSA (FTLD-synuclein). Acta Neuropathol. 2015; 130:299-301.
- Seo JH, Yong SW, Song SK, Lee JE, Sohn YH, Lee PH. A case-control study of multiple system atrophy in Korean patients. Mov Disord. 2010; 25:1953-1959.
- Vanacore N, Bonifati V, Fabbrini G, Colosimo C, De Michele G, Marconi R, et al. Case-control study of multiple system atrophy. Mov Disord. 2005; 20:158-163.
- Hara K, Momose Y, Tokiguchi S, Shimohata M, Terajima K, Onodera O, et al. Multiplex families with multiple system atrophy. Arch Neurol. 2007; 64:545-551.
- Wullner U, Schmitt I, Kammal M, Kretzschmar HA, Neumann M. Definite multiple system atrophy in a German family. J Neurol Neurosurg Psychiatry. 2009; 80:449-450.
- Federoff M, Price TR, Sailer A, Scholz S, Hernandez D, Nicolas A, et al. Genome-wide estimate of the heritability of multiple system atrophy. Parkinsonism Relat Disord. 2016; 22:35-41.
- Chen YP, Zhao B, Cao B, Song W, Guo X, Wei QQ, et al. Mutation scanning of the COQ2 gene in ethnic Chinese patients with multiple-system atrophy. Neurobiol Aging. (2015b); 36:1222 e1227-1211.
- Ogaki K, Fujioka S, Heckman MG, Rayaprolu S, Soto-Ortolaza AI, Labbe C, et al. Analysis of COQ2 gene in multiple system atrophy. Mol Neurodegener. 2014; 9:44.
- Kasai T, Tokuda T, Ohmichi T, Ishii R, Tatebe H, Nakagawa M, et al. Serum levels of coenzyme Q10 in patients with multiple system atrophy. PLoS One. 2016; 11:e0147574.
- Scholz SW, Houlden H, Schulte C, Sharma M, Li A, Berg D, et al. SNCA variants are associated with increased risk for multiple system atrophy. Ann Neurol. 2009; 65:610-614.
- Ross OA, Vilarino-Guell C, Wszolek ZK, Farrer MJ, Dickson DW. Reply to: SNCA variants are associated with increased risk of multiple system atrophy. Ann Neurol. 2010; 67:414-415.
- Sun Z, Xiang X, Tang B, Chen Z, Peng H, Xia K, et al. SNP rs11931074 of the SNCA gene may not be associated with multiple system atrophy in Chinese population. Int J Neurosci. 2015; 125:612-615.
- Ahmed Z, Asi YT, Sailer A, Lees AJ, Houlden H, Revesz T. The neuropathology, pathophysiology and genetics of multiple system atrophy. Neuropathol Appl Neurobiol. 2012; 38:4-24.
- Fujishiro H, Imamura AY, Lin WL, Uchikado H, Mark MH, Golbe LI, et al. Diversity of pathological features other than Lewy bodies in familial Parkinson's disease due to SNCA mutations. Am J Neurodegener Dis. 2013; 2:266-275.
- Mitsui J, Matsukawa T, Sasaki H, Yabe I, Matsushima M, Durr A, et al. Variants associated with Gaucher disease in multiple system atrophy. Ann Clin Transl Neurol. 2015; 2:417-426.
- Srulijes K, Hauser AK, Guella I, Asselta R, Brockmann K, Schulte C, et al. No association of GBA mutations and multiple system atrophy. Eur J Neurol. 2013; 20:e61-62.
- Schottlaender LV, Holton JL, Houlden H. Multiple system atrophy and repeat expansions in c9orf72. JAMA Neurol. 2014; 71:1190-1191.
- Heckman MG, Schottlaender L, Soto-Ortolaza AI, Diehl NN, Rayaprolu S, Ogaki K, et al. LRRK2 exonic variants and risk of multiple system atrophy. Neurology. 2014; 83:2256-2261.
- Scholz SW, Majounie E, Revesz T, Holton JL, Okun MS, Houlden H, et al. Multiple system atrophy is not caused by C9orf72 hexanucleotide repeat expansions. Neurobiol Aging. 2015; 36:1223 e1221-1222.
- Mills JD, Kim WS, Halliday GM, Janitz M. Transcriptome analysis of grey and white matter cortical tissue in multiple system atrophy. Neurogenetics. 2015; 16:107-122.
- Mills JD, Ward M, Kim WS, Halliday GM, Janitz M. Strand-specific RNA-sequencing analysis of multiple system atrophy brain transcriptome. Neuroscience. 2016; 322:234-250.
- Ubhi K, Rockenstein E, Kragh C, Inglis C, Spencer B, Michael S, et al. Widespread microRNA dysregulation in multiple system atrophy - disease-related alteration in miR-96. Eur J Neurosci. 2014; 39:1026-1041.
- Prendecki M, Dorszewska J. The role of microrna in the pathogenesis and diagnosis of neurodegenerative diseases. Austin Alzheimers Parkinsons Dis. 2014; 1:10.
- Chen J, Mills JD, Halliday GM, Janitz M. Role of transcriptional control in multiple system atrophy. Neurobiol Aging. 2015a; 36:394-400.
- Bleasel JM, Wong JH, Halliday GM, Kim WS. Lipid dysfunction and pathogenesis of multiple system atrophy. Acta Neuropathol Commun. 2014; 2:15.
- Kiely AP, Asi YT, Kara E, Limousin P, Ling H, Lewis P, et al. alpha-Synucleinopathy associated with G51D SNCA mutation: a link between Parkinson's disease and multiple system atrophy? Acta Neuropathol. 2013; 125:753-769.
- Kiely AP, Ling H, Asi YT, Kara E, Proukakis C, Schapira AH, et al. Distinct clinical and neuropathological features of G51D SNCA mutation cases compared with SNCA duplication and H50Q mutation. Mol Neurodegener. 2015; 10:41.
- Pasanen P, Myllykangas L, Siitonen M, Raunio A, Kaakkola S, Lyytinen J, et al. A novel alpha-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson's disease-type pathology. Neurobiol Aging. 2014; 35:2180 e2181-2185.
- Sturm E, Stefanova N. Multiple system atrophy: genetic or epigenetic? Exp Neurobiol. 2014; 23:277-291.
- Kuzdas-Wood D, Fellner L, Premstaller M, Borm C, Bloem B, Kirik D, t al. Overexpression of alpha-synuclein in oligodendrocytes does not increase susceptibility to focal striatal excitotoxicity. BMC Neurosci. 2015; 16:86.
- Wenning GK, Jellinger KA. The role of alpha-synuclein in the pathogenesis of multiple system atrophy. Acta Neuropathol. 2005; 109:129-140.
- Jellinger KA. Neuropathology and pathogenesis of multiple system atrophy: an update. Curr Trends Neurol. 2015; 9:45-54.
- Benarroch EE, Schmeichel AM, Low PA, Parisi JE. Differential involvement of the periaqueductal gray in multiple system atrophy. Auton Neurosci. 2010; 158:111-117.
- Ozawa T. Morphological substrate of autonomic failure and neurohormonal dysfunction in multiple system atrophy: impact on determining phenotype spectrum. Acta Neuropathol (Berl). 2007; 114:201-211.
- Stefanova N, Wenning GK. Multiple system atrophy: emerging targets for interventional therapies. Neuropathol Appl Neurobiol; 2016.
- VanderHorst VG, Samardzic T, Saper CB, Anderson MP, Nag S, Schneider JA, et al. alpha-Synuclein pathology accumulates in sacral spinal visceral sensory pathways. Ann Neurol. 2015; 78:142-149.
- Wenning GK, Stefanova N, Jellinger KA, Poewe W, Schlossmacher MG. Multiple system atrophy: a primary oligodendrogliopathy. Ann Neurol. 2008; 64:239-246.
- Jellinger KA, Lantos PL. Papp-Lantos inclusions and the pathogenesis of multiple system atrophy: an update. Acta Neuropathol. 2010; 119:657-667.
- Bleasel JM, Halliday GM, Kim WS. Animal modeling an oligodendrogliopathy - multiple system atrophy. Acta Neuropathol Commun. 2016; 4:12.
- Jellinger KA, Seppi K, Wenning GK. Grading of neuropathology in multiple system atrophy: proposal for a novel scale. Mov Disord. 2005; S29-36.
- Halliday GM, Holton JL, Revesz T, Dickson DW. Neuropathology underlying clinical variability in patients with synucleinopathies. Acta Neuropathol. 2011; 122:187-204.
- Ozawa T. Pathology and genetics of multiple system atrophy: an approach to determining genetic susceptibility spectrum. Acta Neuropathol. 2006; 112:531-538.
- Tong J, Ang LC, Williams B, Furukawa Y, Fitzmaurice P, Guttman M, et al. Low levels of astroglial markers in Parkinson's disease: relationship to alpha-synuclein accumulation. Neurobiol Dis. 2015; 82:243-253.
- Cykowski MD, Coon EA, Powell SZ, Jenkins SM, Benarroch EE, Low PA, et al. Expanding the spectrum of neuronal pathology in multiple system atrophy. Brain. 2015; 138:2293-2309.
- Mendoza-Santiesteban CE, Palma JA, Martinez J, Norcliffe-Kaufmann L, Hedges TR 3rd, Kaufmann H. Progressive retinal structure abnormalities in multiple system atrophy. Mov Disord. 2015; 30:1944-1953.
- Nakamura K, Mori F, Kon T, Tanji K, Miki Y, Tomiyama M, et al. Accumulation of phosphorylated alpha-synuclein in subpial and periventricular astrocytes in multiple system atrophy of long duration. Neuropathology; 2015a.
- Nakamura K, Mori F, Kon T, Tanji K, Miki Y, Tomiyama M, et al. Filamentous aggregations of phosphorylated alpha-synuclein in Schwann cells (Schwann cell cytoplasmic inclusions) in multiple system atrophy. Acta Neuropathol Commun. 2015b; 3:29.
- Kim JS, Yang JJ, Lee DK, Lee JM, Youn J, Cho JW. Cognitive impairment and its structural correlates in the parkinsonian subtype of multiple system atrophy. Neurodegener Dis. 2015b; 15:294-300.
- Cao B, Zhao B, Wei QQ, Chen K, Yang J, Ou R, et al. The Global Cognition, Frontal Lobe Dysfunction and Behavior Changes in Chinese Patients with Multiple System Atrophy. PLoS One. 2015; 10:e0139773.
- Salvesen L, Ullerup BH, Sunay FB, Brudek T, Lokkegaard A, Agander TK, et al. Changes in total cell numbers of the basal ganglia in patients with multiple system atrophy - A stereological study. Neurobiol. Dis, 2015a; 74:104-113.
- Salvesen L, Winge K, Brudek T, Agander TK, Lokkegaard A, Pakkenberg B. Neocortical neuronal loss in patients with multiple system atrophy: a stereological study. Cereb Cortex. 2015b.
- Lee JH, Kim TH, Mun CW, Han YH. Progression of subcortical atrophy and iron deposition in multiple system atrophy: a comparison between clinical subtypes. J Neurol, online. 2015 May 28, 10.1007/s00415-00015-07785-00415.
- Asi YT, Ling H, Ahmed Z, Lees AJ, Revesz T, Holton JL. Neuropathological features of multiple system atrophy with cognitive impairment. Mov Disord. 2014a; 29:884-888.
- Don AS, Hsiao JH, Bleasel JM, Couttas TA, Halliday GM, Kim W. Altered lipid levels provide evidence for myelin dysfunction in multiple system atrophy. Acta Neuropathol Commun. 2014; 2:150.
- Lu CF, Wang PS, Lao YL, Wu HM, Soong BW, Wu YT. Medullo-ponto-cerebellar white matter degeneration altered brain network organization and cortical morphology in multiple system atrophy. Brain Struct Funct. 2014; 219:947-958.
- Indelicato E, Fanciulli A, Poewe W, Antonini A, Pontieri FE, Wenning GK. Cerebral autoregulation and white matter lesions in Parkinson's disease and multiple system atrophy. Parkinsonism Relat Disord. 2015; 21:1393-1397.
- Poston KL, Tang CC, Eckert T, Dhawan V, Frucht S, Vonsattel JP, et al. Network correlates of disease severity in multiple system atrophy. Neurology. 2012; 78:1237-1244.
- Pouclet H, Lebouvier T, Coron E, Rouaud T, Flamant M, Toulgoat F, et al. Analysis of colonic alpha-synuclein pathology in multiple system atrophy. Parkinsonism Relat Disord. 2012; 18: 893-895.
- Provitera V, Nolano M, Caporaso G, Stancanelli A, Manganelli F, Iodice R, et al. Postganglionic sudomotor denervation in patients with multiple system atrophy. Neurology. 2014; 82: 2223-2229.
- Sone M, Yoshida M, Hashizume Y, Hishikawa N, Sobue G. alpha-Synuclein-immunoreactive structure formation is enhanced in sympathetic ganglia of patients with multiple system atrophy. Acta Neuropathol. 2005; 110:19-26.
- Iodice V, Lipp A, Ahlskog JE, Sandroni P, Fealey RD, Parisi JE, et al. Autopsy confirmed multiple system atrophy cases: Mayo experience and role of autonomic function tests. J Neurol Neurosurg Psychiatry. 2012; 83: 453-459.
- Zange L, Noack C, Hahn K, Stenzel, Lipp A. Phosphorylated alpha-synuclein in skin nerve fibres differentiates Parkinson's disease from multiple system atrophy. Brain. 2015; 138:2310-2321.
- Doppler K, Weis J, Karl K, Ebert S, Ebentheuer J, Trenkwalder C, et al. Distinctive distribution of phospho-alpha-synuclein in dermal nerves in multiple system atrophy. Mov Disord. 2015; 30:1688-1692.
- Haga R, Sugimoto K, Nishijima H, Miki Y, Suzuki C, Wakabayashi K, et al. Clinical utility of skin biopsy in differentiating between Parkinson's disease and multiple system atrophy. Parkinsons Dis. 2015; 167038.
- Beyer K, Ariza A. Protein aggregation mechanisms in synucleinopathies: commonalities and differences. J Neuropathol Exp Neurol. 2007; 66:965-974.
- Chavarria C, Souza JM. Oxidation and nitration of alpha-synuclein and their implications in neurodegenerative diseases. Arch Biochem Biophys. 2013;533:25-32.
- Kahle PJ, Neumann M, Ozmen L, Muller V, Jacobsen H, Spooren W, et al. Hyperphosphorylation and insolubility of alpha-synuclein in transgenic mouse oligodendrocytes. EMBO Rep. 2002; 3:583-588.
- Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, et al. alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol. 2002; 4:160-164.
- Hasegawa M, Fujiwara H, Nonaka T, Wakabayashi K, Takahashi H, Lee VM, et al. Phosphorylated alpha-synuclein is ubiquitinated in alpha-synucleinopathy lesions. J Biol Chem. 2002; 277:49071-49076.
- Tong J, Wong H, Guttman M, Ang LC, Forno LS, Shimadzu M, et al. Brain alpha-synuclein accumulation in multiple system atrophy, Parkinson's disease and progressive supranuclear palsy: a comparative investigation. Brain. 2012; 133:172-188.
- Campbell BC, McLean CA, Culvenor JG, Gai WP, Blumbergs PC, Jakala P, et al. The solubility of alpha-synuclein in multiple system atrophy differs from that of dementia with Lewy bodies and Parkinson's disease. J Neurochem.2001; 76:87-96.
- Brudek T, Winge K, Rasmussen NB, Bahl JM, Tanassi J, Agander TK, et al. Altered alpha-synuclein, parkin, and synphilin isoform levels in multiple system atrophy brains. J Neurochem. 2016; 136:172-185.
- Kikuchi A, Takeda A, Okamura N, Tashiro M, Hasegawa T, Furumoto S, et al. In vivo visualization of alpha-synuclein deposition by carbon-11-labelled 2-[2-(2-dimethylaminothiazol-5-yl)ethenyl]-6-[2-(fluoro)ethoxy]benzoxazole positron emission tomography in multiple system atrophy. Brain. 2010; 133:1772-1778.
- Schwarz L, Goldbaum O, Bergmann M, Probst-Cousin S, Richter-Landsberg C. Involvement of macroautophagy in multiple system atrophy and protein aggregate formation in oligodendrocytes. J Mol Neurosci. 2012; 47: 256-266.
- Vallelunga A, Ragusa M, Di Mauro S, Iannitti T, Pilleri M, Biundo R, et al. Identification of circulating microRNAs for the differential diagnosis of Parkinson's disease and Multiple System Atrophy. Front Cell Neurosci. 2014; 8:156.
- Pukass K, Richter-Landsberg C. Inhibition of UCH-L1 in oligodendroglial cells results in microtubule stabilization and prevents alpha-synuclein aggregate formation by activating the autophagic pathway: implications for multiple system atrophy. Front Cell Neurosci. 2015; 9:163.
- Shults CW, Rockenstein E, Crews L, Adame A, Mante M, Larrea G, et al. Neurological and neurodegenerative alterations in a transgenic mouse model expressing human alpha-synuclein under oligodendrocyte promoter: implications for multiple system atrophy. J Neurosci. 2005; 25:10689-10699.
- Ubhi K, Rockenstein E, Mante M, Inglis C, Adame A, Patrick C, et al. Neurodegeneration in a transgenic mouse model of multiple system atrophy is associated with altered expression of oligodendroglial-derived neurotrophic factors. J Neurosci. 2010b; 30:6236-6246.
- Ubhi K, Rockenstein E, Mante M, Inglis C, Adame A, Patrick C, et al. Alpha-synuclein deficient mice are resistant to toxin-induced multiple system atrophy. Neuroreport. 2010a; 21:457-462.
- Wong JH, Halliday GM, Kim WS. Exploring myelin dysfunction in multiple system atrophy. Exp Neurobiol. 2014; 23:337-344.
- Bassil F, Monvoisin A, Canron MH, Vital A, Meissner WG, Tison F, et al. Region-specific alterations of matrix metalloproteinase activity in multiple system atrophy. Mov Disord. 2015; 30:1802-1812.
- Asi YT, Simpson JE, Heath PR, Wharton SB, Lees AJ, Revesz T, et al. Alpha-synuclein mRNA expression in oligodendrocytes in MSA. Glia. 2014b; 62:964-970.
- Djelloul M, Holmqvist S, Boza-Serrano A, Azevedo C, Yeung MS, Goldwurm S, et al. Alpha-synuclein expression in the oligodendrocyte lineage: an in vitro and in vivo study using rodent and human models. Stem Cell Reports. 2015; 5:174-184.
- Ahmed Z, Asi YT, Lees AJ, Revesz T, Holton JL. Identification and quantification of oligodendrocyte precursor cells in multiple system atrophy, progressive supranuclear palsy and Parkinson's disease. Brain Pathol. 2013; 23:263-273.
- Ettle B, Reiprich S, Deusser J, Schlachetzki JC, Xiang W, Prots I, et al. Intracellular alpha-synuclein affects early maturation of primary oligodendrocyte progenitor cells. Mol Cell Neurosci. 2014; 62:68-78.
- May VE, Ettle B, Poehler AM, Nuber S, Ubhi K, Rockenstein E, et al. alpha-Synuclein impairs oligodendrocyte progenitor maturation in multiple system atrophy. Neurobiol Aging. 2014; 35:2357-2368.
- Stefanova N, Reindl M, Neumann M, Haass C, Poewe W, Kahle PJ, et al. Oxidative stress in transgenic mice with oligodendroglial alpha-synuclein overexpression replicates the characteristic neuropathology of multiple system atrophy. Am J Pathol. 2005; 166:869-876.
- Stemberger S, Poewe W, Wenning GK, Stefanova N. Targeted overexpression of human alpha-synuclein in oligodendroglia induces lesions linked to MSA-like progressive autonomic failure. Exp Neurol, 2010; 224:459-464.
- Kuzdas D, Stemberger S, Gaburro S, Stefanova N, Singewald N, Wenning GK. Oligodendroglial alpha-synucleinopathy and MSA-like cardiovascular autonomic failure: experimental evidence. Exp Neurol. 2013; 247:531-536.
- Stefanova N, Wenning GK. Animal models of multiple system atrophy. Clin Auton Res. 2015, 25:9-17.
- Reyes JF, Rey NL, Bousset L, Melki R, Brundin P, Angot E. Alpha-synuclein transfers from neurons to oligodendrocytes. Glia. 2014; 62: 387-398.
- Vieira BD, Radford RA, Chung RS, Guillemin GJ, Pountney DL. Neuroinflammation in multiple system atrophy: response to and cause of alpha-synuclein aggregation. Front Cell Neurosci. 2015; 9:437.
- Kisos H, Pukass K, Ben-Hur T, Richter-Landsberg C, Sharon R. Increased neuronal alpha-synuclein pathology associates with its accumulation in oligodendrocytes in mice modeling alpha-synucleinopathies. PLoS One. 2012; 7:e46817.
- Masuda-Suzukake M, Nonaka T, Hosokawa M, Kubo M, Shimozawa A, Akiyama H, et al. Pathological alpha-synuclein propagates through neural networks. Acta Neuropathol Commun. 2014; 2:88.
- Watts JC, Giles K, Oehler A, Middleton L, Dexter DT, Gentleman SM, et al. Transmission of multiple system atrophy prions to transgenic mice. Proc Natl Acad Sci U S A. 2013; 110:19555-19560.
- Sacino AN, Brooks M, Thomas MA, McKinney AB, McGarvey NH, Rutherford NJ, et al. Amyloidogenic alpha-synuclein seeds do not invariably induce rapid, widespread pathology in mice. Acta Neuropathol. 2014; 127: 645-665.
- Prusiner SB, Woerman AL, Mordes DA, Watts JC, Rampersaud R, Berry DB, et al. (2015). Evidence for alpha-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc Natl Acad Sci U S A, 2015; 112:E5308-5317.
- Peelaerts W, Bousset L, Van der Perren A, Moskalyuk A, Pulizzi R, Giugliano M, et al. alpha-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature. 2015; 522:340-344.
- Rey NL, George S, Brundin P. Review: Spreading the word: precise animal models and validated methods are vital when evaluating prion-like behaviour of alpha-synuclein. Neuropathol Appl Neurobiol. 2016; 42:51-76.
- Peelaerts W, Baekelandt V. a-Synuclein strains and the variable pathologies of synucleinopathies. J Neurochem. 2016.
- Jellinger KA. Multiple system atrophy - a synucleinopathy with specific glioneuronal degeneration. Austin J Clin Neurol. 2015b; 2: 1071.
- Armstrong RA, Cairns NJ, Lantos PL. A quantitative study of the pathological changes in ten patients with multiple system atrophy (MSA). J Neural Transm (Vienna). 2004; 111:485-495.
- Ovadi J, Orosz F. An unstructured protein with destructive potential: TPPP/p25 in neurodegeneration. Bioessays. 2009; 31:676-686.
- Song YJ, Lundvig DM, Huang Y, Gai WP, Blumbergs PC, Hojrup P, et al. p25alpha relocalizes in oligodendroglia from myelin to cytoplasmic inclusions in multiple system atrophy. Am J Pathol. 2007; 171:1291-1303.
- Ota K, Obayashi M, Ozaki K, Ichinose S, Kakita A, Tada M, et al. Relocation of p25 inverted question mark/tubulin polymerization promoting protein from the nucleus to the perinuclear cytoplasm in the oligodendroglia of sporadic and COQ2 mutant multiple system atrophy. Acta Neuropathol Commun. 2014; 2:136.
- Yazawa I, Giasson BI, Sasaki R, Zhang B, Joyce S, Uryu K, et al. Mouse model of multiple system atrophy alpha-synuclein expression in oligodendrocytes causes glial and neuronal degeneration. Neuron. 2005; 45:847-859.
- Kragh CL, Gysbers AM, Rockenstein E, Murphy K, Halliday GM, Masliah E, et al. Prodegenerative IkappaBalpha expression in oligodendroglial alpha-synuclein models of multiple system atrophy. Neurobiol Dis. 2014; 63:171-183.
- von Bernhardi R, Eugenin-von Bernhardi L, Eugenin J. Microglial cell dysregulation in brain aging and neurodegeneration. Front Aging Neurosci. 2015; 7:124.
- Urbizu A, Canet-Pons J, Munoz-Marmol AM, Aldecoa I, Lopez MT, Compta Y, et al. Cystatin C is differentially involved in multiple system atrophy phenotypes. Neuropathol Appl Neurobiol. 2015; 41:507-519.
- Halliday GM. Re-evaluating the glio-centric view of multiple system atrophy by highlighting the neuronal involvement. Brain. 2015; 138:2116-2119.
- Rockenstein E, Ubhi K, Inglis C, Mante M, Patrick C, Adame A, et al. Neuronal to oligodendroglial alpha-synuclein redistribution in a double transgenic model of multiple system atrophy. Neuroreport. 2012; 23:259-264.
- Stefanova N, Georgievska B, Eriksson H, Poewe W, Wenning GK. Myeloperoxidase inhibition ameliorates multiple system atrophy-like degeneration in a transgenic mouse model. Neurotox Res. 2012a; 21:393-404.
- Beraud D, Hathaway HA, Trecki J, Chasovskikh S, Johnson DA, Johnson JA, et al. Microglial activation and antioxidant responses induced by the Parkinson's disease protein alpha-synuclein. J Neuroimmune Pharmacol. 2013; 8:94-117.
- Hayakawa H, Nagai M, Kawanami A, Nakata Y, Nihira T, Ogino M, et al. Loss of DARPP-32 and calbindin in multiple system atrophy. J Neural Transm (Vienna). 2013; 120:1689-1698.
- Stefanova N, Kaufmann WA, Humpel C, Poewe W, Wenning GK. Systemic proteasome inhibition triggers neurodegeneration in a transgenic mouse model expressing human alpha-synuclein under oligodendrocyte promoter: implications for multiple system atrophy. Acta Neuropathol. 2012b; 124:51-65.
- Kawamoto Y, Ito H, Ihara M, Takahashi R. XIAP immunoreactivity in glial and neuronal cytoplasmic inclusions in multiple system atrophy. Clin Neuropathol. 2014; 33:76-83.
- Furukawa Y, Vigouroux S, Wong H, Guttman M, Rajput AH, Ang L, et al. Brain proteasomal function in sporadic Parkinson's disease and related disorders. Ann Neurol. 2002; 51:779-782.
- Tanji K, Odagiri S, Maruyama A, Mori F, Kakita A, Takahashi H, et al. Alteration of autophagosomal proteins in the brain of multiple system atrophy. Neurobiol Dis. 2013; 49:190-198.
- Ubhi K, Low P, Masliah E. Multiple system atrophy: a clinical and neuropathological perspective. Trends Neurosci. 2011; 34:581-590.
- Jellinger KA. What's new in multiple system atrophy. Curr Neurobio. 2015c; 6:11-14.
- Jellinger KA, Krismer F. Aetiopathogenesis. In: Wenning GK, Fanciulli A,editors. Multiple System Atrophy. Vienna: Springer-Verlag; 2014: 57-81
- Kang J-H, Mollenhauer B, Coffey CS, Toledo JB, Weintraub D, Galasko DR, et al. CSF biomarkers associated with disease heterogeneity in early Parkinson disease: the Parkinson's Progression Markers Initiative study. Acta Neuropathol, in print, 2016. doi 10.1007/s00401-00016-01552-00402.
- Simonsen AH, Kuiperij B, El-Agnaf OM, Engelborghs S, Herukka SK, Parnetti L, et al. The utility of alpha-synuclein as biofluid marker in neurodegenerative diseases: a systematic review of the literature. Biomark Med. 2016; 10:19-34.
- Blennow K, Zetterberg H. The past and the future of Alzheimer's disease CSF biomarkers-a journey toward validated biochemical tests covering the whole spectrum of molecular events. Front Neurosci. 2015; 9:345.
- Parnetti L, Cicognola C, Eusebi P, Chiasserini D. Value of cerebrospinal fluid alpha-synuclein species as biomarker in Parkinson's diagnosis and prognosis. Biomark Med. 2016; 10:35-49.
- Delenclos M, Jones DR, McLean PJ, Uitti RJ. Biomarkers in Parkinson's disease: Advances and strategies. Parkinsonism Relat Disord. 2016; 22 Suppl 1:S106-110.
- Algarni MA, Stoessl AJ. The role of biomarkers and imaging in Parkinson's disease. Expert Rev Neurother. 2016; 16:187-203.
- Majbour NK, Vaikath NN, van Dijk KD, Ardah MT, Varghese S, Vesterager LB, et al. Oligomeric and phosphorylated alpha-synuclein as potential CSF biomarkers for Parkinson's disease. Mol Neurodegener. 2016; 11:7.
- Miller DB, O'Callaghan JP. Biomarkers of Parkinson's disease: present and future. Metabolism. 2015; 64:S40-46.
- Brooks DJ, Seppi K. Proposed neuroimaging criteria for the diagnosis of multiple system atrophy. Mov Disord. 2009; 24:949-964.
- Kasahara S, Miki Y, Kanagaki M, Kondo T, Yamamoto A, Morimoto E, et al. "Hot cross bun" sign in multiple system atrophy with predominant cerebellar ataxia: a comparison between proton density-weighted imaging and T2-weighted imaging. Eur J Radiol. 2012; 81:2848-2852.
- Ramli N, Nair SR, Ramli NM, Lim SY. Differentiating multiple-system atrophy from Parkinson's disease. Clin Radiol. 2015; 70:555-564.
- Barbagallo G, Sierra-Pena M, Nemmi F, Traon AP, Meissner WG, Rascol O, et al. Multimodal MRI assessment of nigro-striatal pathway in multiple system atrophy and Parkinson disease. Mov Disord. 2015.
- Sugiyama A, Ito S, Suichi T, Sakurai T, Mukai H, Yokota H, Yonezu T, et al. Putaminal hypointensity on T2*-weighted MR imaging is the most practically useful sign in diagnosing multiple system atrophy: A preliminary study. J Neurol Sci. 2015; 349:174-178.
- Ji L, Wang Y, Zhu D, Liu W, Shi J. White matter differences between multiple system atrophy (parkinsonian type) and Parkinson's disease: A diffusion tensor image study. Neuroscience. 2015; 305:109-116.
- Perju-Dumbrava LD, Kovacs GG, Pirker S, Jellinger K, Hoffmann M, Asenbaum S, et al. Dopamine transporter imaging in autopsy-confirmed Parkinson's disease and multiple system atrophy. Mov Disord. 2012; 27:65-71.
- Scherfler C, Gobel G, Muller C, Nocker M, Wenning GK, Schocke M, et al. Diagnostic potential of automated subcortical volume segmentation in atypical parkinsonism. Neurology. 2016.
- Chung EJ, Lee WY, Yoon WT, Kim BJ, Lee GH. MIBG scintigraphy for differentiating Parkinson's disease with autonomic dysfunction from Parkinsonism-predominant multiple system atrophy. Mov Disord. 2009; 24:1650-1655.
- Laurens B, Constantinescu R, Freeman R, Gerhard A, Jellinger K, Jeromin A, et al. Fluid biomarkers in multiple system atrophy: A review of the MSA Biomarker Initiative. Neurobiol Dis. 2015; 80: 29-41.
- Herbert MK, Aerts MB, Beenes M, Norgren N, Esselink RA, Bloem BR, et al. CSF neurofilament light chain but not FLT3 ligand discriminates Parkinsonian disorders. Front Neurol. 2015; 6:91.
- Herbert MK, Eeftens JM, Aerts MB, Esselink RA, Bloem BR, Kuiperij HB, et al. CSF levels of DJ-1 and tau distinguish MSA patients from PD patients and controls. Parkinsonism Relat Disord. 2014; 20:112-115.
- Shi M, Bradner J, Hancock AM, Chung KA, Quinn JF, Peskind ER, et al. Cerebrospinal fluid biomarkers for Parkinson disease diagnosis and progression. Ann Neurol. 2011; 69:570-580.
- Magdalinou NK, Paterson RW, Schott JM, Fox NC, Mummery C, Blennow K, et al. A panel of nine cerebrospinal fluid biomarkers may identify patients with atypical parkinsonian syndromes. J Neurol Neurosurg Psychiatry. 2015; 86:1240-1247.
- Low PA, Robertson D, Gilman S, Kaufmann H, Singer W, Biaggioni I, et al. Efficacy and safety of rifampicin for multiple system atrophy: a randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2014; 13:268-275.
- Palma JA, Kaufmann H. Novel therapeutic approaches in multiple system atrophy. Clin Auton Res. 2015; 25:37-45.
- Chelban V, Bettencourt C, Houlden H. Updates on potential therapeutic targets in MSA. ACNR. 2016; 15:8-11.
- Poewe W, Seppi K, Fitzer-Attas CJ, Wenning GK, Gilman S, Low PA, et al. Efficacy of rasagiline in patients with the parkinsonian variant of multiple system atrophy: a randomised, placebo-controlled trial. Lancet Neurol. 2015; 14:145-152.
- Peeraully T. Multiple system atrophy. Semin Neurol. 2014; 34:174-181.
- Granata R, Wenning GK. Multiple system atrophy. In: Colosimo C, Riley DE, Wenning GK, editors. Handbook of Atypical Parkinsonism. Cambridge: Cambridge University Press; 2011. p 27-57.
- Mandler M, Valera E, Rockenstein E, Mante M, Weninger H, Patrick C, et al. Active immunization against alpha-synuclein ameliorates the degenerative pathology and prevents demyelination in a model of multiple system atrophy. Mol Neurodegener. 2015; 10:10.
- Spencer B, Valera E, Rockenstein E, Trejo-Morales M, Adame A, Masliah E. A brain-targeted, modified neurosin (kallikrein-6) reduces alpha-synuclein accumulation in a mouse model of multiple system atrophy. Mol Neurodegener. 2015; 10:48.
- Bartus RT, Kordower JH, Johnson EM Jr, Brown L, Kruegel BR, Chu Y, et al. Post-mortem assessment of the short and long-term effects of the trophic factor neurturin in patients with alpha-synucleinopathies. Neurobiol Dis. 2015; 78:162-171.
- Valera E, Masliah E. Combination therapies: The next logical step for the treatment of synucleinopathies? Mov Disord, 2016; 31:225-234.
- Meissner WG, Laurencin C, Tranchant C, Witjas T, Viallet F, Guehl D, et al. Outcome of deep brain stimulation in slowly progressive multiple system atrophy: A clinico-pathological series and review of the literature. Parkinsonism Relat Disord. 2016; 24:69-75.
- Valera E, Monzio Compagnoni G, Masliah E. Review: Novel treatment strategies targeting alpha-synuclein in multiple system atrophy as a model of synucleinopathy. Neuropathol Appl Neurobiol. 2016; 42:95-106.