
Sirtuins and Neurodegeneration
Gizem Yalcin
Department of Medical Biology, Faculty of Medicine, Adnan Menderes University, Aydin, Turkey
Abstract
Sirtuins are highly conserved NAD+-dependent enzymes connected to an increasing set of biological processes. These enzymes have attracted major interest because of their roles in age-related diseases. Sirtuins are implicated in various biological pathways related to stress response, mitochondrial dysfunction, oxidative stress, protein aggregation and inflammatory processes that are intertwined with age-related neurodegenerative diseases.
Introduction
The silencer information regulator (Sir) family of proteins has attracted much attention during the past decade due to its prominent role in many cellular processes. The first sirtuin (Sir2) was discovered in 1984 in yeast1; however, interest did not begin until an effect on life span was observed, in yeast (Saccharomyces cerevisiae)2. Subsequent studies in C. elegans3 and then the fruit fly, D. melanogaster4 proved successful in corroborating the effect of increased lifespan. More specifically, the effect of increased lifespan in the fruit fly was correlated with calorie restriction (CR). Afterwards, it was shown that Sir2 extends longevity minimally at best in C. elegans and the role of Sir2 in D. melanogaster is controversial5. Sirtuins were recognized as NAD+-dependent deacetylases6, and then implicated in chromatin silencing and the metabolic pathways7. Once the enzymatic functions of sirtuins were elucidated, sirtuins ability to extend life span were shown to involve similar pathways as those of CR. However, the literature regarding the role of yeast, fly and worm sirtuins in CR is controversial. On the other hand, mammalian sirtuins were found to have various functions in the central nervous system, liver, pancreas, skeletal muscle, and adipose tissue8.
There are seven homologs (SIRT1-7) with different enzymatic activities and localizations in the mammalian cell. SIRT1, -6, and -7 reside primarily in the nucleus and have direct effects on nuclear transcription of genes involved in metabolism; however SIRT1 shuttles to the cytoplasm when required to act on cytoplasmic targets9. SIRT1 is highly expressed in the adult brain with high levels in the cortex, hippocampus, cerebellum and hypothalamus9. There are lower levels of SIRT1 in white matter10. SIRT1 is primarily expressed in neurons and considered a nuclear protein11,12. However, studies have shown that it is also located in cytoplasm9. SIRT2 is cytoplasmic and deacetylates tubulin microtubules and transcription factors that shuttle from the cytoplasm to the nucleus13. SIRT2 is expressed primarily in oligodendrocytes and plays a role in myelin sheath formation as well as oligodendroglial differentiation14. SIRT3-5 resides in the mitochondrial matrix and regulates various enzymes in the tricarboxylic acid and urea cycles, oxidative phosphorylation and reactive oxygen species production12. One study suggests that SIRT3 is a nuclear NAD+-dependent histone deacetylase that translocates to the mitochondria upon cellular stress15. SIRT6 and SIRT7 are localized to the nucleus, being present in heterochromatin and nucleoli, respectively12. A recent study showed that SIRT6 regulates lifespan in male mice16.
SIRT1’s role in Neurodegeneration
SIRT1 is the most extensively studied sirtuin among all sirtuins. SIRT1 was subsequently studied as a mediator of CR, a dietary regimen aimed at reducing caloric intake by 20-30% without malnutrition. CR was previously shown to slow down the aging process, increasing health and lifespan in several animal models17-22. A recent study demonstrated that hypothalamic overexpression of SIRT1 extends longevity and delays aging in mice23. These phenotypes of mice are achieved by increased neural activity observed in the dorsomedial and lateral hypothalamic nuclei.
Aging is the major risk factor for the development of neurodegenerative diseases. SIRT1 orchestrates different stress response pathways. More explicitly, SIRT1 targets multiple transcriptional regulators, such as p5324, FOXO325 and NFκB26. Since SIRT1 is also important in regulating mitochondrial function, it is expected that some of the protective roles of SIRT1 against neurodegenerative diseases might be through its effects in mitochondria.
SIRT1’s role in Alzheimer’s Disease
Aging population carries a significant amount of neurodegenerative disease, due to increased life expectancy. The most common serious neurodegenerative disorder is Alzheimer’s disease (AD) causing severe cognitive and behavioral deficits. AD affects 35 million people worldwide and is the leading cause of dementia among the elderly27. Aβ is the product of the proteolytic cleavage of the amyloid-β protein precursor (AβPP) into 38, 40, or 42 amino acid peptides and is viewed as the toxic mediator of AD28. Aβ oligomerization is toxic to neurons in vitro and in vivo and produces synaptic dysfunction, calcium dysregulation, oxidative stress, and neuroinflammation29. Moreover, Aβ (specifically Aβ42) is the primary component of the senile plaques that develop in the brains of AD30.
The mounting evidence for the implications of the effects of SIRT1 in AD initially came from research primarily focused on the effects of SIRT1 overexpression mediated by small molecule modulators, such as NAD+ or resveratrol. For example, in vitro, both were found to reduce Aβ oligomers in a concentration dependent manner32. In addition, both small molecule modulators were found to ameliorate oxidative stress33-35. Another study with SIRT1 overexpression found that Aβ toxicity in microglia was reduced via inhibition of NFκB signaling36 that induces inflammation.
CR was shown to attenuate Alzheimer's disease type brain amyloidosis in Squirrel monkeys37 and was subsequently studied in the APPswe/PSEN1dE9 mouse line35. The research suggested that attenuation of beta-amyloid content in the brain during CR can be reproduced in mouse neurons in vitro by manipulating cellular SIRT1 expression/activity through mechanisms involving the regulation of the serine/threonine Rho kinase ROCK1, known in part for its role in the inhibition of the non-amyloidogenic α-secretase processing of the APP. These results demonstrated for the first time a role for SIRT1 in the brain as a novel mechanism through which CR may influence AD amyloid neuropathology.
In an in vitro work using N2a cells expressing human Swedish APP mutation (N2aSwe), it was shown that overexpressing SIRT1 gene resulted in a significant increase in ADAM10 and sAPPa levels in this cell type31. Cilostazol (OPC-13013, 6-[4-(1-cyclohexyl-1H-te- trazol-5-yl) butoxy]-3,4-dihydro-2-[1H]-quinolinone) was previously shown to increase intracellular cyclic AMP (cAMP) levels via inhibiting type III phosphodiesterase enzyme46. In this study, cilostazol was demonstrated to enhance the protein expressions of RARb and ADAM10. In addition, ADAM10 elevation induced by cilastazol was significantly attenuated by LE135 (a RARb inhibitor), sirtinol (SIRT1 inhibitor), and RARb-gene silencing. The authors conclude that the increase in the levels of ADAM10/a-secretase activity and the reduction in the intracellular Ab accumulation, which are based on the cilostazol-stimulated SIRT1-linked RARb activation, ameliorate AD-associated neurodegeneration31. In N2aSwe cells, increased Ab accumulation was accompanied by increased Ac-tau and P-tau levels together with elevated P300 and GSK3b P-Tyr216 expression; their expressions were significantly reduced by cilostazol and resveratrol treatments38. This study showed that increased cAMP-dependent protein kinase-linked CK2/ SIRT1 expression by cilostazol may suppress the tau-related neurodegeneration38.
Another study demonstrated that apolipoprotein E ε4 allele (ApoE) significantly reduces the ratio of soluble amyloid precursor protein alpha (sAPPa) to Ab and also decreases SirT1 expression39. It was also shown that Ab reduces SIRT1 expression, triggers Tau phosphorylation and APP phosphorylation. Additionally, programmed cell death is induced as a result of Ab accumulation39. Therapeutic up-regulation of SIRT1 might provide opportunities for the amelioration of Alzheimer's-disease-type neuropathology through inhibition of amyloidogenesis. Ultimately, further analysis into this aspect is necessary if any progress is to be made.
Other transgenic mice studies include a p25 transgenic mouse, which overexpresses the cdk5-activating human p25 protein thereby exhibiting tau hyperphosphorylation and neurodegeneration with features similar to AD40. Tau is a microtubule-binding protein found in high content in neurons and is responsible for the assembly and stability of microtubules. Hyperphosphorylation of the tau protein leads toward tangles in an ordered fashion such as paired helices and straight filaments. These self-assembled tangles are implicated in the pathogenesis of AD as well as other taupathies. Cyclin-dependent kinase 5 (cdk5) and its regulatory subunit p35 have important roles in the development of the mammalian central nervous system38. Proteolytic cleavage of p35 yields p25, which then activates cdk5. Accumulation of p25 is a hallmark of neurodegenerative disease. In a mouse model (cdk5-activating p25), injection of the SIRT1-activating polyphenol resveratrol resulted in less hippocampal degeneration, less cognitive deficit, and reduced acetylation of SIRT1 substrates PGC-1α and p5341. Additionally, when SIRT1 was directly overexpressed in the hippocampus of these mice using a lentiviral vector, the effects of resveratrol injections were corroborated.
Tau and SIRT1 have previously shown a relationship in that tau is acetylated at multiple lysine residues and SIRT1 regulates the level of phosphorylated tau via deacetylation42. When tau is acetylated by the histone acetyltransferase, p300, the breakdown of tau is inhibited. SIRT1 inhibition was shown to increase tau levels thereby increasing phosphorylated tau. In summary, degradation of phosphorylated tau improved cognitive function and reduced neuronal cell death39.
SIRT1’role in Parkinson’s disease
Parkinson’s disease (PD) is another progressive neurodegenerative disorder affecting the central nervous system. The neuropathology involves loss of dopaminergic neurons in the substantia nigra pars compacta, and its symptoms include muscle rigidity, bradykinesia, resting tremor and postural instability, amongst others. Cytoplasmic inclusions called Lewy bodies (LB) containing the protein α-synuclein43, some proteasomal and lysosomal subunits have been found on histological analysis of PD. Although the reason for the neuronal cell loss still remains elusive, misfolding, oligomerization and α-synuclein aggregation have been implicated in the neuropathology of PD and parkinsonian plus disorders such as multiple systems atrophy (MSA)44. Studies of PD models and the functions of genes implicated in inherited forms of PD have looked at two different possibilities leading toward dopaminergic neuronal cell death: (i) mitochondrial dysfunction and oxidative stress, and (ii) misfolding and aggregation of proteins45.
In other cell culture studies, dietary resveratrol was shown to attenuate oxidative stress induced by the parkinsonian mimetic 6-hydroxydopapamine46 as well as the controversial treatment for PD, L-3,4-dihydroxyphenylalanine (L-dopa)47. The authors of these studies suggest that the antioxidant properties of resveratrol are involved in its attenuation of oxidative stress as the neuroprotective effect, rather than investigating SIRT1 activation. Moreover, resveratrol exhibited a neuroprotective effect on dopaminergic neurons in midbrain slice culture from multiple insults48. In this study, it was determined that the neuroprotective effect was indeed, void of SIRT1 activation because SIRT1 inhibitors did not attenuate the protective resveratrol effects.
Studies performed so far suggests that SIRT1 may be a pharmacological target in the treatment of neurodegenerative diseases by deacetylating different target proteins. However, additional studies are essential to substantially prove SIRT1 provides a molecular basis of protection. For example, one study, which used the well-established 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) model of PD in SIRT1 transgenic mice, resulted in no protection49. Neuron-specific enolase (NSE) SIRT1 transgenic mice were generated to overexpress human SIRT1 in neurons. Overexpression of SIRT1 did not have any neuroprotective effects against the neuronal damage induced by ischemia or MPTP48.
SIRT1’s role in Huntington’s Disease
Huntington's disease (HD) is a neurodegenerative disease that affects four to seven individuals per 100,000. HD is a fatal neurodegenerative disorder caused by an expanded polyglutamine repeat in huntingtin (HTT) protein.
In a mouse model of HD, brain-specific knockout of SIRT1 results in exacerbation of brain pathology, whereas overexpression of SIRT1 improved survival, neuropathology and the expression of BDNF51. SIRT1 deacetylase activity was shown to directly target neurons to mediate neuroprotection from mutant HTT. The neuroprotective effect of SIRT1 requires the presence of CREB-regulated transcription coactivator 1 (CRTC1), a brain-specific modulator of CREB activity. Under normal conditions, SIRT1 deacetylates and activates CRTC1 by promoting its dephosphorylation and its interaction with CREB. BDNF was identified as a key target of SIRT1 and CRTC1 transcriptional activity in both normal and HD neurons. Mutant HTT interferes with the CRTC1-CREB interaction to repress BDNF transcription, and SIRT1 rescues this defect in vitro and in vivo50. Together, these findings show a neuroprotective role for SIRT1 in mammalian HD models52.
SIRT1’s role in ALS
Loss of neurons with age is also seen in amyotrophic lateral sclerosis (ALS). The underlying causes of denervation of the neuromuscular junction and eventual motor neuron death in ALS have not been resolved.
The superoxide dismutase 1 (SOD1)(G93A) mutant mouse is a frequently used animal model of ALS. As mentioned earlier, resveratrol, a polyphenolic molecule that enhances SIRT1 activity, improved motor function and survival in the SOD1 mouse model via modulation of p53 acetylation38. Manipulation of SIRT1 deacetylase activity had effects at the protein level in healthy aging organisms; however, resveratrol treatment did not lead to functional improvement or increased longevity in a mouse model of ALS53. In a similar mouse model, region specific changes in the immunoreactivity of SIRT1 expression in the central nervous system was observed. SIRT1 increases in cerebral cortical pyramidal cells, hippocampal pyramidal cells of area CA1-3 and dentate gyrus cells, thalamus, and spinal cord54.
The role that SIRT1 activation plays in the pathogenesis of ALS remains unclear; however, via activating SIRT1, resveratrol was shown to protect against neurodegeneration in a cell model of ALS55. SIRT1 expression was found to be much lower in the mutant (SOD1)(G93A)-bearing VSC4.1 cells compared to (SOD1)(wt) cells when both were cultured in low-serum medium, indicating the involvement of SIRT1 activation defects in the pathogenesis of ALS under energetic stress. Further investigation revealed that resveratrol had a dose-dependent protective effect on this ALS cell model. This further demonstrated a role for SIRT1 activation in the protection of neuronal cells from degeneration. These findings suggest that resveratrol can protect the ALS cell model from mutant SOD1-mediated toxicity through up-regulating the expression of SIRT1, which might represent a potential therapeutic target for preventing the motor neuron degeneration in ALS patients.
SIRT2’s role in Neurodegeneration
SIRT2 is an oligodendroglial cytoplasmic protein and localized to the outer and juxtanodal loops in the myelin sheath. Among cytoplasmic proteins of OLN-93 oligodendrocytes, alpha-tubulin is the main substrate of SIRT2 deacetylase14. There exists a counterbalancing role of SIRT2 against a facilitator effect of tubulin acetylation on oligodendroglial differentiation. SIRT2 availability to oligodendroglia has important implications for myelinogenesis, myelin-axon interaction, and brain aging. In addition to α-tubulin and histone H4 substrates, SIRT2 deacetylates forkhead transcription factors of class O, FOXO1 and FOXO314. Since FOXO transcription factors are involved in several cellular processes, including yet not limited to DNA repair, cell cycle, apoptosis, metabolism and aging, SIRT2 has been initially investigated in these processes56. The effective control of FOXO activity in response to environmental stimuli is likely to be critical to prevent aging and age-dependent diseases, including cancer, neurodegenerative diseases and diabetes.
SIRT2’s role in Parkinson’s Disease
SIRT2 inhibition, pharmacologically or genetically, was found to be beneficial in the rescue of alpha-synuclein toxicity in both in vitro and in vivo models of Parkinson’s Disease (PD). A potent inhibitor of SIRT2 was shown to inhibit alpha-synuclein toxicity and modified inclusion morphology in a cellular model of PD57. Genetic inhibition of SIRT2 via small interfering RNA similarly rescued alpha-synuclein toxicity. In addition, the inhibitors protected against dopaminergic cell death both in vitro and in a Drosophila model of PD. In addition, the interaction between oligomeric alpha-synuclein and acetylated microtubules was investigated as a source of neurodegeneration. SIRT2 inhibition was shown to increase microtubule-dependent transport of neurotoxic alpha-synuclein oligomers to the nucleation aggregation site, facilitating formation of inclusions called LB57.
It was recently shown that SIRT2 in the brain enhanced MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine)-induced nigrostriatal damage via deacetylating FOXO3a and activating Bim59. MPTP is a dopaminergic neurotoxin that replicates most of the clinical features of PD and produces a reliable and reproducible lesion of the nigrostriatal dopaminergic pathway and neurodegeneration after its systemic administration57. Chronic administration of MPTP induces lesion via apoptosis57. It was shown that SIRT2 deacetylates Foxo3a, increases RNA and protein levels of Bim, a proapoptotic factor, and as a result, enhances apoptosis in the MPTP model of PD. Neurodegeneration induced by chronic MPTP regimen is prevented by genetic deletion of SIRT2 in mouse. Deletion of SIRT2 lead to the reduction of apoptosis due to an increase in acetylation of Foxo3a and a decrease in Bim levels59.
SIRT2’s role in Huntington’s Disease
The molecular pathogenesis of Huntington’s disease (HD) is complex and many mechanisms and cellular processes have been proposed as potential sites of therapeutic intervention. Previous studies in invertebrate and cell culture HD models have suggested that inhibition of SIRT2 could have beneficial consequences on disease progression60,61. SIRT2 has been proposed to deacetylate α-tubulin, histone H4 K16 and to regulate cholesterol biogenesis, a pathway, which is dysregulated in HD patients and HD mouse models58. The distribution and function of the SIRT2 microtubule deacetylase in differentiated, postmitotic neurons were investigated and it was determined that expression of specific isoforms of SIRT2 in the mammalian central nervous system exhibits age-dependent accumulation in the mouse brain and spinal cord. Furthermore, endogenous SIRT2 expression correlates with reduced α-tubulin acetylation in primary mouse cortical neurons; therefore, it is postulated that brain-enriched species of SIRT2 may function as the microtubule deacetylases in mature neurons62.
An in vivo study, SIRT2 was reduced or ablated to further explore the function of SIRT2 and to assess whether SIRT2 loss has a beneficial impact on disease progression in the R6/2 mouse model of HD63. The reduction or loss of SIRT2 had no effect on the acetylation of α-tubulin or H4K16 or on cholesterol biosynthesis in the brains of wild type mice. In addition, genetic reduction or ablation of SIRT2 had no effect on HD progression as assessed by a battery of physiological and behavioral tests. Therefore, it was concluded that SIRT2 inhibition does not modify disease progression in the R6/2 mouse model of HD and SIRT2 inhibition should not be prioritized as a therapeutic option for HD60.
Another in vivo model utilizes the efficacy of a brain-permeable SIRT2 inhibitor in two separate genetic mouse models of HD. It was reported that compound treatment resulted in improved motor function, extended survival, and reduced brain atrophy with reduction of aggregated mutant huntingtin (a hallmark of HD pathology)64. In addition, genetic or pharmacologic inhibition of SIRT2 in a striatal neuronal model of HD resulted in gene expression changes including significant down-regulation of the RNAs responsible for sterol biosynthesis61. Mutant huntingtin fragments increases sterols in neuronal cells; therefore, manipulation of sterol biosynthesis at the transcriptional level would prove beneficial.
Most of the studies showed that the inhibition of SIRT2 is beneficial for HD except the study that used R6/2 as a model60. R6/2 mouse model displays aggressive HD phenotype, therefore additional studies should be designed where different HD mouse models are utilized. The outcome of the deletion of SIRT2 in these mouse models should be analyzed in order to assess the role of SIRT2 in HD.
In summary, in all the disease models studied, inhibition of SIRT2 seems to have beneficial effects by deacetylating different targets. Therefore, inhibiting the activity of this enzyme might be effective in designing effective therapies.
Mitochondrial Sirtuins and Neurodegeneration
Three of the mammalian sirtuins, SIRT3, SIRT4 and SIRT5 are localized in mitochondria, which are the major organelles responsible for energy production, balance and metabolism65. SIRT3 was shown to have strong deacetylase activity. On the other hand, SIRT4 has adenosine diphosphate (ADP)–ribosylation activity, and SIRT5 has desuccinylase and demalonylase activity in addition to weak deacetylation activity65. A recent study showed that SIRT3 localizes to the mitochondria, does not exist in nucleus and it is a proteolytically modified deacetylase66. The study also emphasizes that SIRT3 does the majority of deacetylation in mitochondria66,67. A number of targets were identified for SIRT3; however, only a handful of substrates were described for SIRT4 and SIRT5. Mitochondrial dysfunction is associated with age-related disorders such as metabolic syndrome, cancer and neurodegeneration. The role of mitochondrial sirtuins in brain and their relation to neurodegeneration is not broadly studied except for a few recent in vivo studies using toxins such as kainic acid and MPTP.
SIRT3 was shown to attenuate MPTP-induced nigrostriatal damage in mice68. SIRT3 KO mice displayed increased degeneration compared to wild type mice after MPTP administration. Decreased levels of superoxide dismutase 2 (SOD2), a specific mitochondrial antioxidant enzyme, and glutathione peroxidase expression was detected in MPTP-induced SIRT3 KO mice compared to wild-type controls68. Therefore, it was concluded that SIRT3 might be protective in MPTP-induced neurodegeneration via improving antioxidant capacity in mitochondria68. A similar study was conducted using SIRT5 KO mice. MPTP-induced nigrostriatal degeneration was elevated in SIRT5 KO mice together with a larger decrease in the expression level of SOD2 compared to wild type controls69.
SIRT4 was shown to have different physiological roles in various tissues but its role in brain was unexplored. A recent study showed that SIRT4 is up-regulated in brain in response to treatment with kainic acid, an excitotoxin70. Glutamate transport keeps low extracellular levels of glutamate in the brain to prevent excitotoxicity that leads to neurodegeneration and inefficient neurotransmission. SIRT4 KO mice displayed severe seizure phenotypes compared to wild type mice after exposure to kainic acid70. Deletion of SIRT4 also leads to reduced glutamate transporter expression in brain after kainic acid administration. This study showed a novel stress response role for SIRT4 in maintaining efficient glutamate transport and preventing excitotoxicity70.
SIRT6, SIRT7 and Neurodegeneration
SIRT6 is a nuclear enzyme known to deacetylate histone H3-lysine 9 (H3K9). In previous studies, SIRT6 was found to promote longevity71, 72. Due to the phenotype observed in SIRT6 knockout mice, this protein is also thought to regulate DNA base excision repair (BER) and metabolic functions72. A more recent study showed that SIRT6 regulates the stability of Tau protein and the lack of SIRT6 increases hyperphosrylated tau levels73. Moreover, RNA and protein levels of SIRT6 were found to be decreased in Alzheimer’s disease patients. Therefore, SIRT6 and its downstream targets could be targeted for therapeutical strategies in neurodegenerative diseases.
SIRT7 is the least characterized sirtuin and found in nucleolus74. It interacts with rDNA and regulates RNA Pol I74. SIRT7 was shown to be overexpressed in many human tumors75 and recent studies identified SIRT7 as a proto-oncogene76. However, SIRT7 is largely unexplored for neurodegenerative diseases and brain. Similar expression could be modulated in cell and animal models of neurodegenerative diseases, similar to the previous studies conducted for other sirtuins. SIRT7 is also expected to have major functions in neuronal pathways and diseases.
Conclusion
The functions of sirtuins in the brain are still unknown; however, published studies show that they have important roles in neurodegenerative diseases. Substrates of sirtuins are molecules that take part in the fundamental cellular pathways and are crucial to the biology of the organism. Investigating the roles of sirtuins in neurodegeneration not only elucidates whether these molecules are of importance therapeutically but also identifies their novel functions in brain (Figure 1).
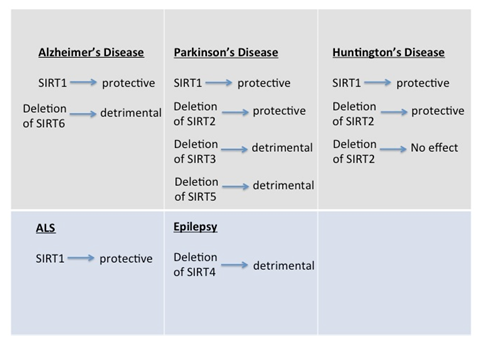
Figure 1: Summary of the effects of sirtuins on neurodegenerative diseases
Although all seven mammalian sirtuins are expressed in the brain, only the roles of SIRT1 and SIRT2 in neurodegeneration were extensively studied (Figure 1). The findings show that SIRT1 activation generally displays a protective role against neurodegenerative diseases; whereas, SIRT2 inhibition or deletion shows a positive outcome. These two enzymes seem to be quite different although they share a common catalytic domain. They have different targets and they are located in different subcellular locations. In light of the studies conducted, we can conclude that activating SIRT1 and inhibiting SIRT2 seem to be beneficial for the organism. On the other hand, deletion of mitochondrial sirtuins in mice display increased degeneration in brain after administration of toxins. Therefore, designing pharmacological activators or inhibitors for sirtuins is a crucial research area since they might be beneficial against neurodegenerative diseases. It is extremely important to develop selective activators or inhibitors that target a specific sirtuin since they have different functions in different cellular compartments. Additionally, the level of sirtuins in specific areas of the brain may hold a prognostic value for the detection or diagnosis of the diseases. However, the best modulators of sirtuins against age-related disorders can only be designed if we understand the molecular mechanisms underlying their effects in neurodegenerative diseases and brain.
References
- Shore D, Squire M, Nasmyth KA. Characterization of two genes required for the position-effect control of yeast mating-type genes. Embo J. 1984; 3: 2817-2823.
- Kaeberlein M, McVey M, Guarente L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 1999; 13: 2570-2580.
- Tissenbaum HA, Guarente L. Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature. 2001; 410: 227-230.
- Rogina B, Helfand SL. Sir2 mediates longevity in the fly through a pathway related to calorie restriction. Proc Natl Acad Sci U S A. 2004; 101: 15998-16003.
- Burnett C, Valentini S, Cabreiro F, et al. Absence of effects of Sir2 overexpression on lifespan in C. elegans and Drosophila. Nature. 2011; 477: 482-485.
- Imai S, Armstrong CM, Kaeberlein M, et al. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000; 403: 795-800.
- Guarente L. Sir2 links chromatin silencing, metabolism, and aging. Genes Dev. 2000; 14: 1021-1026.
- Donmez G, Guarente L. Aging and disease: connections to sirtuins. Aging Cell. 2010; 9: 285-290.
- Tanno M, Sakamoto J, Miura T et al. Nucleocytoplasmic shuttling of the NAD+-dependent histone deacetylase SIRT1. J Biol Chem. 2007; 282: 6823-6832.
- Ramadori G, Lee CE, Bookout AL, et al. Brain SIRT1: anatomical distribution and regulation by energy availability. J Neurosci. 2008; 28: 9989-9996.
- Sakamoto J, Miura T, Shimamoto K, et al. Predominant expression of Sir2alpha, an NAD-dependent histone deacetylase, in the embryonic mouse heart and brain. FEBS Lett. 2004; 556: 281-286.
- Michishita E, Park JY, Burneskis JM, et al. Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Mol Biol Cell. 2005; 16: 4623-4635.
- North BJ, Marshall BL, Borra MT, et al. The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol Cell. 2003; 11: 437-444.
- Li W, Zhang B, Tang J, et al. Sirtuin 2, a mammalian homolog of yeast silent information regulator-2 longevity regulator, is an oligodendroglial protein that decelerates cell differentiation through deacetylating alpha-tubulin. J Neurosci. 2007; 27: 2606-2616.
- Scher MB, Vaquero A, Reinberg D. SirT3 is a nuclear NAD+-dependent histone deacetylase that translocates to the mitochondria upon cellular stress. Genes Dev. 2007; 21: 920-928.
- Kanfi Y, Naiman S, Amir G, et al. The sirtuin SIRT6 regulates lifespan in male mice. Nature. 2012; 483: 218-221.
- Weindruch R, Walford RL. Dietary restriction in mice beginning at 1 year of age: effect on life-span and spontaneous cancer incidence. Science. 1982; 215: 1415-1418.
- Austad SN. Life extension by dietary restriction in the bowl and doily spider, Frontinella pyramitela. Exp Gerontol. 1989; 24: 83-92.
- Jiang JC, Jaruga E, Repnevskaya MV, et al. An intervention resembling caloric restriction prolongs life span and retards aging in yeast. Faseb J. 2000; 14: 2135-2137.
- Mair W, Goymer P, Pletcher SD, et al. Demography of dietary restriction and death in Drosophila. Science. 2003; 301: 1731-1733.
- Lakowski B, Hekimi S. The genetics of caloric restriction in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 1998; 95: 13091-13096.
- Bishop NA, Guarente L. Genetic links between diet and lifespan: shared mechanisms from yeast to humans. Nat Rev Genet. 2007; 8: 835-844.
- Satoh A, Brace CS, Ben-Josef G, et al. SIRT1 promotes the central adaptive response to diet restriction through activation of the dorsomedial and lateral nuclei of the hypothalamus. J Neurosci. 2010 Jul 28; 30(30): 10220-32.
- Langley E, Pearson M, Faretta M, et al. Human SIR2 deacetylates p53 and antagonizes PML/p53-induced cellular senescence. Embo J. 2002; 21: 2383-2396.
- Brunet A, Sweeney LB, Sturgill JF, et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004; 303: 2011-2015.
- Yeung F, Hoberg JE, Ramsey CS, et al. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. Embo J. 2004; 23: 2369-2380.
- Castellani RJ, Rolston RK, Smith MA. Alzheimer disease. Dis Mon. 2010; 56: 484-546.
- Hardy JA, Higgins GA. Alzheimer's disease: the amyloid cascade hypothesis. Science. 1992; 256: 184-185.
- Walsh DM, Selkoe DJ. A beta oligomers - a decade of discovery. J Neurochem. 2007; 101: 1172-1184.
- Querfurth HW, LaFerla FM. Alzheimer's disease. N Engl J Med. 2010; 362: 329-344.
- Lee HR, Shin HK, Park SY, et al. Cilostazol suppresses b-amyloid production by activating a disintegrin and metalloproteinase 10 via the upregulation of SIRT1-coupled Retinoic Acid Receptor b. J Neuroscience Res. 2014; 92: 1581-1590.
- Wang J, Fivecoat H, Ho L, et al. The role of Sirt1: at the crossroad between promotion of longevity and protection against Alzheimer's disease neuropathology. Biochim Biophys Acta. 2010; 1804: 1690-1694.
- Albani D, Polito L, Batelli S, et al. The SIRT1 activator resveratrol protects SK-N-BE cells from oxidative stress and against toxicity caused by alpha-synuclein or amyloid-beta (1-42) peptide. J Neurochem. 2009; 110: 1445-1456.
- Feige JN, Lagouge M, Canto C, et al. Specific SIRT1 activation mimics low energy levels and protects against diet-induced metabolic disorders by enhancing fat oxidation. Cell Metab. 2008; 8: 347-358.
- Qin W, Yang T, Ho L, et al. Neuronal SIRT1 activation as a novel mechanism underlying the prevention of Alzheimer disease amyloid neuropathology by calorie restriction. J Biol Chem. 2006; 281: 21745-21754.
- Chen J, Zhou Y, Mueller-Steiner S, et al. SIRT1 protects against microglia-dependent amyloid-beta toxicity through inhibiting NF-kappaB signaling. J Biol Chem. 2005; 280: 40364-40374.
- Qin W, Chachich M, Lane M, et al. Calorie restriction attenuates Alzheimer's disease type brain amyloidosis in Squirrel monkeys (Saimiri sciureus). J Alzheimers Dis. 2006; 10: 417-422.
- Lee HR, Shin HK, Park SY, et al. Attenuation of b-amyloid-induced taupathy via activation of CK2a/SIRT1: targeting for cilostazol. J Neurosci Res. 2014; 92: 206-217.
- Theendakara V, Patent A, Peters Libeu CA, et al. Neuroprotective Sirtuin ratio reversed by ApoE4. Proc Natl Acad Sci U S A. 2013; 110: 18303-183008.
- Cruz JC, Tseng HC, Goldman JA, et al. Aberrant Cdk5 activation by p25 triggers pathological events leading to neurodegeneration and neurofibrillary tangles. Neuron. 2003; 40: 471-483.
- Kim D, Nguyen MD, Dobbin MM, et al. SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer's disease and amyotrophic lateral sclerosis. Embo J. 2007; 26: 3169-3179.
- Min SW, Cho SH, Zhou Y, et al. Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron. 2010; 67: 953-966.
- Spillantini MG, Schmidt ML, Lee VM, et al. Alpha-synuclein in Lewy bodies. Nature. 1997; 388: 839-840.
- Outeiro TF, Marques O, Kazantsev A. Therapeutic role of sirtuins in neurodegenerative disease. Biochim Biophys Acta. 2008; 1782: 363-369.
- Dauer W, Przedborski S. Parkinson's disease: mechanisms and models. Neuron. 2003; 39: 889-909.
- Chao J, Yu MS, Ho YS, et al. Dietary oxyresveratrol prevents parkinsonian mimetic 6-hydroxydopamine neurotoxicity. Free Radic Biol Med. 2008; 45: 1019-1026.
- Peritore CS, Ho A, Yamamoto BK, et al. Resveratrol attenuates L-DOPA-induced hydrogen peroxide toxicity in neuronal cells. Neuroreport. 2012; 23: 989-994.
- Okawara M, Katsuki H, Kurimoto E, et al. Resveratrol protects dopaminergic neurons in midbrain slice culture from multiple insults. Biochem Pharmacol. 2007; 73: 550-560.
- Kakefuda K, Fujita Y, Oyagi A, et al. Sirtuin 1 overexpression mice show a reference memory deficit, but not neuroprotection. Biochem Biophys Res Commun. 2009; 387: 784-788.
- Sorolla MA, Nierga C, Rodriguez-Colman MJ, et al. Sir2 is induced by oxidative stress in a yeast model of Huntington disease and its activation reduces protein aggregation. Arch Biochem Biophys. 2011; 510: 27-34.
- Jeong H, Cohen DE, Cui L, et al. Sirt1 mediates neuroprotection from mutant huntingtin by activation of the TORC1 and CREB transcriptional pathway. Nat Med. 2012; 18: 159-165.
- Zhang F, Wang S, Gan L, et al. Protective effects and mechanisms of sirtuins in the nervous system. Prog Neurobiol. 2011; 95: 373-395.
- Markert CD, Kim E, Gifondorwa DJ, et al. A single-dose resveratrol treatment in a mouse model of amyotrophic lateral sclerosis. J Med Food. 2010; 13: 1081-1085.
- Lee JC, Shin JH, Park BW, et al. Region-specific changes in the immunoreactivity of SIRT1 expression in the central nervous system of SOD1(G93A) transgenic mice as an in vivo model of amyotrophic lateral sclerosis. Brain Res. 2012; 1433: 20-28.
- Wang J, Zhang Y, Tang L, et al. Protective effects of resveratrol through the up-regulation of SIRT1 expression in the mutant hSOD1-G93A-bearing motor neuron-like cell culture model of amyotrophic lateral sclerosis. Neurosci Lett. 2011; 503: 250-255.
- Calnan DR, Brunet A. The FoxO code. Oncogene. 2008; 27: 2276-2288.
- Outeiro TF, Kontopoulos E, Altmann SM, et al. Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson's disease. Science. 2007; 317: 516-519.
- Alim MA, Ma QL, Takeda K, et al. Demonstration of a role for alpha-synuclein as a functional microtubule-associated protein. J Alzheimers Dis. 2004; 6: 435-442; discussion 443-439.
- Liu L, Arun A, Ellis L, et al. Sirtuin 2 (SIRT2) enhances 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced nigrostriatal damage via apoptotic pathway. Front Aging Neurosci. 2014 6: 184.
- Pallos J, Bodai L, Lukacsovich T, et al. Inhibition of specific HDACs and sirtuins suppresses pathogenesis in a Drosophila model of Huntington's disease. Hum Mol Genet. 2008; 17: 3767-3775.
- Luthi-Carter R, Taylor DM, Pallos J, et al. SIRT2 inhibition achieves neuroprotection by decreasing sterol biosynthesis. Proc Natl Acad Sci U S A. 2010; 107: 7927-7932.
- Maxwell MM, Tomkinson EM, Nobles J, et al. The Sirtuin 2 microtubule deacetylase is an abundant neuronal protein that accumulates in the aging CNS. Hum Mol Genet. 2011; 20: 3986-3996.
- Bobrowska A, Donmez G, Weiss A, et al. SIRT2 ablation has no effect on tubulin acetylation in brain, cholesterol biosynthesis or the progression of Huntington's disease phenotypes in vivo. PLoS One. 2012; 7: e34805.
- Chopra V, Quinti L, Kim J, et al. The sirtuin 2 inhibitor AK-7 is neuroprotective in Huntington's disease mouse models. Cell Rep. 2012; 2: 1492-1497.
- Shih J, Donmez G. Mitochondrial sirtuins as therapeutic targets for age-related disorders. Genes Cancer. 2013; 4(3-4): 91-6.
- Cooper HM, Spelbrink JN. The human SIRT3 protein deacetylase is exclusively mitochondrial. Biochemical Journal. 2008; 411: 279-285.
- Hallows WC, Albaugh BC, Denu JM. Where in the cell is SIRT3?: Functional localization of an NAD+-dependent protein deacetylase. Biochemical Journal. 2008; 411(2): e11-e13.
- Liu L, Peritore C, Ginsberg J, et al. SIRT3 attenuates MPTP-induced nigrostriatal degeneration via enhancing mitochondrial antioxidant capacity. Neurochem Res. 2015; DOI 10.1007/s11064-014-1507-8.
- Liu L, Peritore C, Ginsberg J, et al. Protective role of SIRT5 against motor deficit and dopaminergic degeneration in MPTP-induced mice model of Parkinson's Disease. Behav Brain Res. 2015; 281: 215-21.
- Shih J, Liu L, Mason A, et al. Loss of SIRT4 decreases GLT-1-dependent glutamate uptake and increases sensitivity to kainic acid. J Neurochem. 2014; 131(5): 573-81.
- Liszt G, Ford E, Kurtev M, et al. Mouse Sir2 homolog SIRT6 is a nuclear ADP-ribosyltransferase. J Biol Chem. 2005; 280(22): 21313-20.
- Mostoslavsky R, Chua KF, Lombard DB, et al. Genomic Instability and Aging-like Phenotype in the Absence of Mammalian SIRT6. Cell. 2006; 124: 315–329.
- Kaluski S, Portillo M, Besnard A, et al. Neuroprotective Functions for the Histone Deacetylase SIRT6. Cell Rep. 2017; 18(13): 3052-3062.
- Ford E, Voit R, Liszt G, et al. Mammalian Sir2 homolog SIRT7 is an activator of RNA polymerase I transcription. Genes & Dev. 2006; 20: 1075–1080.
- Ashraf N, Zino S, Macintyre A, et al. Altered sirtuin expression is associated with node-positive breast cancer. Br J Cancer. 2006 Oct 23; 95(8): 1056-61.
- Barber MF, Michishita-Kioi E, Xi Y, et al. SIRT7 links H3K18 deacetylation to maintenance of oncogenic transformation. Nature. 2012; 487(7405): 114-8.