
Amylin Signaling in Diabetes and Alzheimer's Disease: Therapy or Pathology?
John Grizzanti1, Rachel Corrigan1, Spencer Servizi1, Gemma Casadesus1,2*
1School of Biomedical Sciences, Kent State University, Ohio, USA
2Department of Biological Sciences, Kent State University, Ohio, USA
Abstract
Growing evidence highlights the intimate relationship between type II diabetes (T2D) and Alzheimer’s disease (AD). Importantly, these two diseases share a number of pathological similarities, including amyloid accumulation, oxidative stress, inflammation, and cell death. To date, drug therapies for AD and T2D are lacking and there is a crucial need for the discovery and development of novel therapeutics for these diseases. A number of human and rodent studies have given evidence that metabolic hormone supplementation is highly valuable for improving cognitive function and overall metabolic health in both T2D and AD. The pancreatic hormone amylin has arisen as a crucial component of the disease etiology of both T2D and AD, though the exact role that amylin plays in these diseases is not yet well understood. Here, we critically review the current literature that utilizes human amylin or its synthetic analogue, pramlintide, as well as amylin receptor antagonists for the treatment of AD.
Introduction
Alzheimer’s disease (AD) is a progressive, debilitating neurodegenerative disease characterized by the accumulation of amyloid-beta (Aβ) plaques and neurofibrillary tangles composed of hyperphosphorylated tau1. The accumulation of these pathological peptides contributes to deficits in executive functions such as learning and memory, mood, affect, etc. and presents a substantial burden to the patient and caregivers. The incidence of AD is increasing at an alarming rate in the U.S., with an estimated 5.5 million Americans living with AD as of 2017 and this number is expected to triple by 20502. Furthermore, the cost of caring for and treating AD patients currently exceeds $200 billion annually and is only expected to increase3. While AD is clearly a monumental problem within the U.S. and beyond, treatment options remain very limited4. Many drug trials have been conducted with a wide array of targeted approaches, yet there are currently only six drugs approved by the FDA for AD and are only symptomatic treatments5, 6. To date, the majority of pharmacological agents developed have specifically targeted the hallmark Aβ or tau pathology, yet none have been successful in clearing or preventing pathology4. As such, there is a fundamental need to develop viable therapeutics and preventative treatments for AD.
Age-related (sporadic) AD is a complicated multifactoral disease, having numerous genetic and environmental influences. Environment and lifestyle are heavily implicated in the development of sporadic AD; factors such as diet7-9, obesity8-10, metabolic syndrome7, type II diabetes (T2D)9, 11, and cardiovascular disease12 have all been implicated in the causation of AD. Of critical importance, rates of obesity and diabetes are rapidly rising in parallel with AD12, 13. Though the relationship between obesity and AD is somewhat unclear, there is evidence that mid-life obesity plays a role in the development of AD10. More importantly, obesity is commonly accompanied by a number of other diseases, including cardiovascular disease, hypertension, dyslipidemia, T2D, stroke, etc.14. The incidence of T2D is quickly rising, with the CDC estimating that approximately 30.3 million people (1 in 10 adults) in the U.S. have diabetes and a staggering 84.1 million (1 in 3 adults) have prediabetes, most of whom are unaware of their condition. Furthermore, due to a large-scale decrease in physical activity that is accompanied by a simultaneous increase in food intake and poor diet, rates of obesity, T2D, metabolic syndrome, and cardiovascular disease are only proposed to increase to an estimated 600 million T2D cases worldwide by 203515.
The body of evidence implicating metabolic function and disease in the process of cognitive decline and aging is substantial16, 17. For example, approximately 70% of people diagnosed with T2D report cognitive impairment and a substantial number of T2D patients later develop dementia16, 18-21. Individuals diagnosed with T2D for at least five years have a significantly increased risk of developing AD compared to those whom have suffered from T2D for less than five years17. Together these data suggest that the increasing prevalence of T2D in the population may be contributing to the rising rates of AD.
T2D is initially characterized by high blood glucose and insulin, which leads to hyperinsulinemia; importantly, amylin, a small metabolic hormone produced by β-islet cells of the pancreas, is co-packaged and co-secreted with insulin and is thus overproduced in T2D.22. Importantly, there are a number of pathological features that are present in both T2D and AD: 1) decreased brain metabolism and metabolic hormone resistance 2) amyloid pathology 3) oxidative stress (OS) and inflammation. Chronic, hyperinsulinemia and hyperamylinemia leads to a number of physiological issues: chronic hyperinsulinemia leads to system insulin resistance22, impaired insulin transport across the blood-brain barrier (BBB)23, 24, and thus decreased insulin signaling within the brain25. Loss of insulin signaling in the brain is associated with a number of AD-related pathological features, including increased Aβ production, tau phosphorylation, and neuroinflammation.
Furthermore, amylin shares similar pathological features with Aβ at high concentrations26 and may be a common pathway between the two diseases. For example, amylin fibrils have been found in the pancreas of 95% of T2D patients27-29 and cause a number of physiological disruptions including aberrant Ca2+ influx, increased secretion of pro-inflammatory cytokines30,31, and ultimately β-islet cell loss32. Furthermore, amylin readily crosses the BBB and forms amylin fibrils as well as mixed plaques with Aβ within the brain and may be responsible for AD-like pathology and Aβ seeding in T2D33-35. Amylin is known to affect long-term potentiation (LTP) in the hippocampus and may have an innate influence on cognitive function within the brain36-39. However, whether amylin is a toxic insult in these diseases or whether its functional loss through aggregation or late stage β-cell loss in T2D contributes to the development of an AD remains unclear.
The Amylin Signaling Dichotomy
There is still much debate about the involvement of the amylin receptor (AMYR) and amylin signaling in the disease progression and etiology of T2D and AD. The body of research aimed at discerning this relationship is quickly expanding. All relevant research has consistently demonstrated that modulation of amylin signaling affects AD-related pathology. The nature of this relationship, however, has yet to be concretely elucidated. Several groups have produced compelling data suggesting that amylin signaling is beneficial in preventing AD-related pathology and cognitive deficits both in vivo and in vitro40-44. Importantly, pramlintide, a recombinant non-aggregating form of amylin, used in conjunction with insulin therapies to treat diabetes and improves glycemic control, reduces body weight, and reduces serum markers of OS45-47 also shows promise as an AD therapeutic. To date, however, there have been no clinical trials that have aimed to utilize amylin or pramlintide as a therapeutic agent in treating dementia. Clear evidence from rodent studies suggests that chronic treatment with either human amylin or pramlintide poses strong therapeutic benefit in reducing AD-related pathology; amylin/pramlintide supplementation reduces soluble Aβ levels, plaque burden, tau phosphorylation, neuroinflammation, and OS while also improving cognition40-42,44. The above data suggest that a loss of innate amylin signaling in the CNS due to aggregation gives rise to an increased risk for the development of AD and is covered in more detail in Grizzanti et al. 201848.
In contrast, studies also show that human amylin and Aβ have similar toxic effects and that these toxic effects can be alleviated using AMYR antagonist36-39,49. For example, data show that in vivo treatment with AMYR antagonists yields very similar physiological benefits to amylin or pramlintide treatment. Treatment of TgCRND8 AD mice with AC253, an AMYR antagonist, or its cyclic counterpart cAC253 reduces neuroinflammation, soluble Aβ levels, and plaque burden while also improving cognition50. Similarly, in vitro/ex-vivo studies show that low dose human amylin or Aβ causes disruptions in LTP and that these deficits are blocked by AC253 or pramlintide38,39, and higher doses of human amylin/amylin oligomers are associated with uncontrolled Ca2+ influx, which is strongly linked to cell death26,32. Together, these data support a toxic function of amylin oligomers and thus a potential therapeutic mechanism for AMYR blockade. In contrast, others have shown that the beneficial effects of amylin can be blocked using AC25341. Thus, the therapeutic potential of amylin treatment or inhibition remains unclear and highlights the complex and dichotomous nature of amyloids in the brain and periphery.
Piecing Together the Puzzle
There are a number of holes in the current literature that need filling to give a more complete picture of the amylin story: 1) the nature of the innate amylin system and amylin signaling within the brain 2) Aβ and pramlintide signaling capabilities through the three main AMYR and related receptors 3) the therapeutic mechanisms by which amylin/pramlintide or AMYR inhibition are mediated. First, interesting novel data demonstrate that the AMYR is not only involved in signaling, but also in ligand transport across the BBB. The AMYR is a heterodimeric receptor that is composed of a calcitonin receptor and a receptor activity modifying protein (1-3)51. To this end, a 50% global knockdown of the calcitonin receptor (a key component of the AMYR) significantly reduced the amount of AC253 found in the brain50, indicating that AMYR located in the BBB are involved in transporting these ligands into the brain and may also be involved in shuttling amylin and pramlintide into/out of the brain. The existence of these BBB transport mechanisms suggests that amylin likely has innate physiological function in the brain, as its transport into the brain is tightly controlled. However, how amylin signaling or lack thereof leads to the pathological features of AD and whether the AMYR is the vehicle through which Aβ mediates its toxic effects still remains unclear.
Next, conflicting evidence exists with regard to the relationship between Aβ and the AMYR. Though several studies clearly demonstrate that human amylin and Aβ have similar effects on LTP in the CNS and use of AMYR inhibitors ameliorates these deleterious effects36-39, other evidence suggests that Aβ (1-42) is incapable of signaling through the any of the AMYR to evoke any sort of cAMP response at a wide variety of concentrations52. It is possible that Aβ activates different signaling cascades through interaction with the AMYR or simply acts as an inert competitive inhibitor, but this has yet to be demonstrated.
Furthermore, a separate study demonstrated that oligomeric amylin mediates its toxic effects directly through the AMYR and indirectly through TRPV4, a nonselective cation channel26. Low concentrations to of human amylin evoke a Ca2+ response that is mediated through its native receptor. However, at higher concentrations, human amylin forms oligomers and actives aberrant signaling that results in the activation of TRVP4 channels and allows for uncontrolled cation influx, particularly Ca2+. Pharmacological blockade of the AMYR and TRPV4 demonstrates that both receptors are necessary for oligomeric human amylin to induce its toxic Ca2+ effects26. As such, it is likely that Aβ mediates its toxic effects on the AMYR in a similar fashion, though these data do not yet exist. Uncontrolled Ca2+ influx is linked to a number of pathological phenomena, including uncontrolled vesicular release, OS and mitochondrial dysfunction, apoptosis, etc. To this end, it is likely that cellular dysfunction and the development of additional AD-like pathology that arises from toxic amyloid signaling is mediated through both the AMYR and TRPV4. As such, it is necessary to discern the signaling cascades that modulate the relationship between the AMYR and TRVP4. Furthermore, pharmacological experiments are warranted that utilize Aβ and pramlintide over a wide array of doses to determine Aβ and pramlintide’s effects on Ca2+ currents, LTP, cAMP production, and other signaling cascades to determine their signaling capabilities. These experiments will help to fill some of the voids in the current literature with regard to the AMYR and its involvement in disease states (Figure 1).
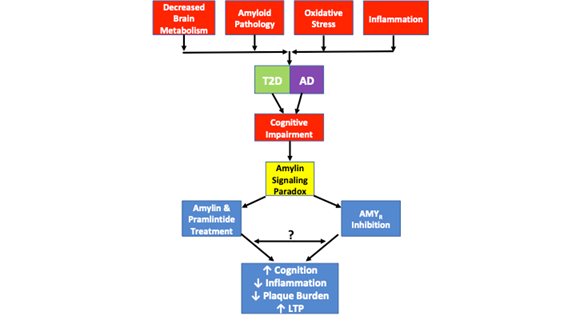
Figure 1. depicts the amylin signaling paradox and pathological similarities observed in T2D and AD. Decreased brain metabolism, amyloid pathology, oxidative stress and inflammation are all common pathological features observed in both diseases. While not every case of T2D or AD includes each of these pathological features, every case exhibits cognitive impairment. The amylin signaling paradox comes in to play as studies have shown that both AMYR inhibition and AMYR agonism via amylin and pramlintide treatment results in improved cognition, decreased inflammation, decreased plaque burden and increased LTP. The signaling mechanisms governed by AMYR agonism and AMYR antagonism have yet to be fully elucidated. While amylin signaling is traditionally associated with cAMP and PKA signaling, it is unclear if other cascades are also activated by amylin/pramlintide. Furthermore, it is unclear if AMYR antagonists, amylin oligomers, or Aβ signal through the AMYR or whether there are any similarities or crosstalk between all of these AMYR ligands. As such, a number of experiments proposed in this review will help to further elucidate the true nature of the AMYR.
Conclusions
The current disparity with regard to the role of amylin signaling in the brain demonstrates an essential need for further elucidation of amylin’s involvement in both AD and T2D. In T2D, it is likely that in the early stages of the disease, amylin floods the brain, forms oligomers, induces aberrant signaling through its native receptor, and recruits TRPV4 to induce pathological Ca2+ influx that results in widespread neuronal dysfunction that manifests as OS, uncontrolled vesicular release and interneuronal dysfunction, inflammation, and resulting cell death. This mechanism may be responsible for the initial transition from the healthy brain to brain aging in metabolic disease. As such, AMYR or TRVP4 inhibition at certain time points in metabolic disease and in the early stages of diabetes may be warranted to block the toxic effects of oligomeric amylin or Aβ. However, strong evidence also suggests that amylin replacement with either human amylin or pramlintide reduces most of the major AD related pathology while also improving cognition in rodent models of AD. As such, amylin signaling replacement with amylin or pramlintide in the mid to late stages of diabetes when amylin signaling is lost due to aggregation, oligomerization, or β-cell loss may be warranted. To this end, there is also a need to discern the temporal presentation of pathological events in metabolically linked brain aging and therapeutic options for early, intermediate, and late stage disease. Critical analysis and testing of the direct nature and signaling capabilities of these amyloids as well as the therapeutic nature of specific temporal treatments may help to bridge the gap between AMYR inhibition therapies and amylin replacement therapies.
Funding
Funding for this article was provided by the National Institutes of Aging grant 1R15AG050292-01A1.
References
- LaFerla FM, Oddo S. Alzheimer's disease: Aβ, tau and synaptic dysfunction. Trends Mol Med. 2005; 11: 170-176.
- Hebert LE, Weuve J, Scherr PA, et al. Alzheimer disease in the United States (2010–2050) estimated using the 2010 census. Neurology. 2013; 80: 1778-1783.
- Association As. 2016 Alzheimer's disease facts and figures. Alzheimer's & Dementia. 2016; 12: 459-509.
- Cummings JL, Morstorf T, Zhong K. Alzheimer’s disease drug-development pipeline: few candidates, frequent failures. Alzheimers Res Ther. 2014; 6: 37.
- Hyde C, Peters J, Bond M, et al. Evolution of the evidence on the effectiveness and cost-effectiveness of acetylcholinesterase inhibitors and memantine for Alzheimer's disease: systematic review and economic model. Age Ageing. 2012; 42: 14-20.
- Howard R, McShane R, Lindesay J, et al. Donepezil and memantine for moderate-to-severe Alzheimer's disease. N Engl J Med. 2012; 366: 893-903.
- Crane PK, Walker R, Hubbard RA, et al. Glucose levels and risk of dementia. N Engl J Med. 2013; 369: 540-548.
- Loef M, Walach H. Midlife obesity and dementia: meta?analysis and adjusted forecast of dementia prevalence in the United States and China. Obesity. 2013; 21.
- Gudala K, Bansal D, Schifano F, et al. Diabetes mellitus and risk of dementia: a meta?analysis of prospective observational studies. Journal of diabetes investigation. 2013; 4: 640-650.
- Whitmer RA, Gunderson EP, Barrett-Connor E, et al. Obesity in middle age and future risk of dementia: a 27 year longitudinal population based study. BMJ. 2005; 330: 1360.
- Vagelatos NT, Eslick GD. Type 2 diabetes as a risk factor for Alzheimer's disease: the confounders, interactions, and neuropathology associated with this relationship. Epidemiol Rev. 2013; 35: 152-160.
- Debette S, Seshadri S, Beiser A, et al. Midlife vascular risk factor exposure accelerates structural brain aging and cognitive decline. Neurology. 2011; 77: 461-468.
- Ginter E, Simko V. Global prevalence and future of diabetes mellitus In Diabetes Springer. 2013; 35-41.
- Khaodhiar L, McCowen KC, Blackburn GL. Obesity and its comorbid conditions. Clin Cornerstone. 1999; 2: 17-31.
- Federation ID. IDF diabetes atlas. Brussels: International Diabetes Federation. 2013.
- K Dash S. Cognitive impairment and diabetes. Recent Pat Endocr Metab Immune Drug Discov. 2013; 7: 155-165.
- Leibson CL, Rocca WA, Hanson V, et al. Risk of dementia among persons with diabetes mellitus: a population-based cohort study. Am J Epidemiol. 1997; 145: 301-308.
- Biessels GJ, Strachan MW, Visseren FL, et al. Dementia and cognitive decline in type 2 diabetes and prediabetic stages: towards targeted interventions. The lancet Diabetes & endocrinology. 2014; 2: 246-255.
- Akter K, Lanza EA, Martin SA, et al. Diabetes mellitus and Alzheimer's disease: shared pathology and treatment. Br J Clin Pharmacol. 2011; 71: 365-376.
- Akter S, Rahman MM, Abe SK, et al. Prevalence of diabetes and prediabetes and their risk factors among Bangladeshi adults: a nationwide survey. Bull World Health Organ. 2014; 92: 204-213A.
- Ott A, Stolk R, Van Harskamp F, et al. Diabetes mellitus and the risk of dementia The Rotterdam Study. Neurology. 1999; 53: 1937-1937.
- Farris W, Mansourian S, Chang Y, et al. Insulin-degrading enzyme regulates the levels of insulin, amyloid β-protein, and the β-amyloid precursor protein intracellular domain in vivo. Proceedings of the National Academy of Sciences. 2003; 100: 4162-4167.
- Schwartz MW, Figlewicz DF, Kahn SE, et al. Insulin binding to brain capillaries is reduced in genetically obese, hyperinsulinemic Zucker rats. Peptides. 1990; 11: 467-472.
- Wallum B, Taborsky Jr G, Porte Jr D, et al. Cerebrospinal Fluid Insulin Levels Increase During Intravenous Insulin Infusions in Man*. The Journal of Clinical Endocrinology & Metabolism. 1987; 64: 190-194.
- Gil-Bea FJ, Solas M, Solomon A, et al. Insulin levels are decreased in the cerebrospinal fluid of women with prodomal Alzheimer's disease. J Alzheimers Dis. 2010; 22: 405-413.
- Zhang N, Yang S, Wang C, et al. Multiple target of hAmylin on rat primary hippocampal neurons. Neuropharmacology. 2017; 113: 241-251.
- Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. 2006; 444: 840-846.
- Johnson K, O'Brien T, Jordan K, et al. Impaired glucose tolerance is associated with increased islet amyloid polypeptide (IAPP) immunoreactivity in pancreatic beta cells. The American journal of pathology. 1989; 135: 245.
- Johnson KH, O'Brien TD, Betsholtz C, et al. Islet amyloid, islet-amyloid polypeptide, and diabetes mellitus. N Engl J Med. 1989; 321: 513-518.
- Masters SL, Dunne A, Subramanian SL, et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1β in type 2 diabetes. Nat Immunol. 2010; 11: 897.
- Wang X, Bao W, Liu J, et al. Inflammatory markers and risk of type 2 diabetes: a systematic review and meta-analysis. Diabetes Care2013; 36: 166-175.
- Kawahara M, Kuroda Y, Arispe N, et al. Alzheimer's β-amyloid, human islet amylin, and prion protein fragment evoke intracellular free calcium elevations by a common mechanism in a hypothalamic GnRH neuronal cell line. J Biol Chem. 2000; 275: 14077-14083.
- Verma N, Ly H, Liu M, et al. Intraneuronal Amylin Deposition, Peroxidative Membrane Injury and Increased IL-1β Synthesis in Brains of Alzheimer’s Disease Patients with Type-2 Diabetes and in Diabetic HIP Rats. J Alzheimers Dis. 2016; 53: 259-272.
- Jackson K, Barisone GA, Diaz E, et al. Amylin deposition in the brain: a second amyloid in Alzheimer disease. Ann Neurol. 2013; 74: 517-526.
- Oskarsson ME, Paulsson JF, Schultz SW, et al. In vivo seeding and cross-seeding of localized amyloidosis: a molecular link between type 2 diabetes and Alzheimer disease. The American journal of pathology. 2015; 185: 834-846.
- Fu W, Patel A, Jhamandas JH. Amylin receptor: a common pathophysiological target in Alzheimer’s disease and diabetes mellitus. Front Aging Neurosci. 2013; 5.
- Fu W, Ruangkittisakul A, MacTavish D, et al. Amyloid β (Aβ) peptide directly activates amylin-3 receptor subtype by triggering multiple intracellular signaling pathways. J Biol Chem. 2012; 287: 18820-18830.
- Kimura R, MacTavish D, Yang J, et al. Pramlintide Antagonizes Beta Amyloid (Aβ)-and Human Amylin-Induced Depression of Hippocampal Long-Term Potentiation. Mol Neurobiol. 2017; 54: 748-754.
- Kimura R, MacTavish D, Yang J, et al. Beta amyloid-induced depression of hippocampal long-term potentiation is mediated through the amylin receptor. J Neurosci. 2012; 32: 17401-17406.
- Zhu H, Wang X, Wallack M, et al. Intraperitoneal injection of the pancreatic peptide amylin potently reduces behavioral impairment and brain amyloid pathology in murine models of Alzheimer's disease. Mol Psychiatry. 2015; 20: 252.
- Zhu H, Xue X, Wang E, et al. Amylin receptor ligands reduce the pathological cascade of Alzheimer's disease. Neuropharmacology. 2017; 119: 170-181.
- Adler BL, Yarchoan M, Hwang HM, et al. Neuroprotective effects of the amylin analogue pramlintide on Alzheimer's disease pathogenesis and cognition. Neurobiol Aging. 2014; 35: 793-801.
- Zhang Z, Hartmann H, Do VM, et al. Destabilization of β-catenin by mutations in presenilin-1 potentiates neuronal apoptosis. Nature. 1998; 395: 698-702.
- Wang E, Zhu H, Wang X, et al. Amylin Treatment Reduces Neuroinflammation and Ameliorates Abnormal Patterns of Gene Expression in the Cerebral Cortex of an Alzheimer’s Disease Mouse Model. J Alzheimers Dis. 2017; 56: 47-61.
- Singh-Franco D, Robles G, Gazze D. Pramlintide acetate injection for the treatment of type 1 and type 2 diabetes mellitus. Clin Ther. 2007; 29: 535-562.
- Singh?Franco D, Perez A, Harrington C. The effect of pramlintide acetate on glycemic control and weight in patients with type 2 diabetes mellitus and in obese patients without diabetes: a systematic review and meta?analysis. Diabetes, Obesity and Metabolism. 2011; 13: 169-180.
- Hollander PA, Levy P, Fineman MS, et al. Pramlintide as an adjunct to insulin therapy improves long-term glycemic and weight control in patients with type 2 diabetes. Diabetes Care. 2003; 26: 784-790.
- Grizzanti J, Corrigan R, Casadesus G. Neuroprotective Effects of Amylin Analogues on Alzheimer’s Disease Pathogenesis and Cognition. J Alzheimers Dis. 2018; 1-13.
- Jhamandas JH, Li Z, Westaway D, et al. Actions of β-amyloid protein on human neurons are expressed through the amylin receptor. The American journal of pathology. 2011; 178: 140-149.
- Soudy R, Patel A, Fu W, et al. Cyclic AC253, a novel amylin receptor antagonist, improves cognitive deficits in a mouse model of Alzheimer's disease. Alzheimer's & Dementia: Translational Research & Clinical Interventions. 2017; 3: 44-56.
- Gebre-Medhin S, Mulder H, Zhang Y, et al. Reduced nociceptive behavior in islet amyloid polypeptide (amylin) knockout mice. Mol Brain Res. 1998; 63: 180-183.
- Gingell JJ, Burns ER, Hay DL. Activity of pramlintide, rat and human amylin but not Aβ1–42 at human amylin receptors. Endocrinology. 2014; 155: 21-26.