
Overview of Promising Rat Models for Cortical Lesion Research- 2006 Until Now
Michaela T. Haindl1, Muammer Ücal2, Franz Fazekas1, Sonja Hochmeister1*
1Department of Neurology, Medical University Graz, Auenbruggerplatz 22, 8036 Graz, Austria
2Research Unit of Experimental Neurotraumatology, Department of Neurosurgery, Medical University Graz, Auenbruggerplatz 29, 8036 Graz, Austria
Abstract
Success in developing new drugs for diseases often depends on laboratory research usually involving animal models. For the early disease phases of multiple sclerosis (MS) this prerequisite was given by experimental autoimmune encephalomyelitis (EAE), an animal model which reflects the pathophysiological mechanisms of the disease quite well. Only a few models resemble cellular features of the progressive disease form of MS (like cortical demyelination) and if they do observable lesions are rather sparse and short lived. The lack of suitable animal models delayed and complicated drug development. Recently a few promising animal models reassembling many characteristics of progressive MS have been described aiming at both a better understanding of cellular mechanisms of this disease phase as well as the development of new therapeutic options. The authors of new rat models postulate novel findings of cortical pathology summarized in this article. The models differ in their focus and not all distinct models are reflecting all features of progressive MS but together they allow research on different aspects of the disease and offer the opportunity to expand our knowledge and clear the way for drug development.
Introduction
Multiple sclerosis (MS) is the most common demyelinating disease of the central nervous system and affects mainly young adults. Disease onset and disease course are highly variable but most patients start with a relapsing-remitting (RR) disease course characterized by alternating demyelination followed by subsequent recovery. After this RR disease phase many of the patients enter a secondary progressive (SP) disease phase, with a steady worsening of clinical neurological symptoms. In contrast to the RR disease phase where new gadolinium-enhancing lesions1 dominate, mainly diffuse white matter abnormalities, atrophy, and cortical demyelination appear in the progressive phase. Only a small subgroup of patients develops primary progressive (PP) MS from the disease onset2. Inflammation is a crucial process in all forms of MS beginning with peripheral immune reaction, migration of immune cells through the blood-brain-barrier (BBB) leading to demyelination and degeneration in white and gray matter. However, this does not exclude the possibility that neurodegeneration can exist independent of inflammation as cortical MS lesions are frequently devoid of profound inflammatory infiltration3,4. In comparison to the RRMS where drug development succeeded over the past 20 years, there are still only two compounds (ocrelizumab and siponimod) available for the progressive disease phase5. Anti-inflammatory and immunomodulatory treatments are beneficial in RRMS but not in progressive disease phases. In this stage, the inflammation is discussed as being trapped behind a closed BBB where those drugs might not reach this inflammatory process3.
Pathogenesis of the Progressive Disease Phase
The pathogenesis of brain damage differs between relapsing and progressive disease phases. In progressive MS new white matter and gadolinium-enhancing lesions are rare; rather the whole brain is affected including global inflammation, extensive cortical demyelination and diffuse axonal injury in the normal appearing white matter (NAWM)6. There are three patterns of cortical demyelination described in the literature based on the location of the lesion. Type 1 lesions are contiguous with subcortical white matter lesions, Type 2 are small lesions confined to the cortex and Type 3 lesions extending from the pial surface to the cortical layer7. Compared to white matter lesions, cortical lesions contain fewer CD3-positive lymphocytes and CD68-positive macrophages7. Also, a remarkable loss of neurons is detectable in the demyelinated cortex7, which is attributed either to direct oxidative injury or retrograde neurodegeneration due to axon injury in the white matter8.
Meningeal inflammation has recently been identified to contribute to the pathogenesis in the progressive disease phase as well. Especially tertiary lymphoid-like structures in SPMS in the leptomeninges composed of T-cells, B-cells and plasma cells with a network of follicular dendritic cells have been proposed to provide a steady inflammatory environment and to contribute to neuronal pathology in the grey matter and permanent clinical disability9. In PPMS no tertiary lymphoid-like structures appear, but diffuse meningeal inflammation and the associated inflammatory milieu in subarachnoid compartments plays a role in the pathogenesis of cortical grey matter lesions and the increased rate of clinical progression10.
Importance of Animal Models for Fundamental Research
A better insight into pathomechanisms behind MS degeneration is crucial for drug development strategies11. No single animal model can mimic all features of human demyelinating diseases, but the available models reflect specific facets crucial for fundamental research. A very common animal model reflecting RRMS pathomechanisms quite well is the experimental autoimmune encephalomyelitis (EAE). It can be reliably induced in a number of different species by active immunization with myelin components. There are different types of EAE available using different myelin preparations with slightly different impact on cellular changes. In general, the spinal cord and cerebellar white matter is mainly affected by EAE. This model already helped to define important disease mechanisms and allowed specific drug development12. Another animal model for studying white matter lesions is the cuprizone model, a toxic demyelination model. This model is commonly induced in mice; for the introduction in the rat contradicting research data is available. Demyelination (of the cerebellum) is induced by feeding milled chow containing cuprizone13. There are also several virus induced models of demyelination available also resembling parts of cellular mechanisms of MS quite well14. For studying MS related mechanisms in the rat however, EAE remains the most popular and frequently used animal model.
Research of the pathogenesis of cortical lesion formation has been limited by the fact that conventional animal models do not regularly affect the cortex15. For testing novel therapeutic agent on progressive pathology and to clarify the pathological mechanisms underlying this disease form animal models are indispensable. For the progressive MS animal models were long discussed as inefficient and too far away from the pathophysiological changes in humans16. But in the last few years, several excellent rat models have been developed trying to close this missing gap15,17–19. In this mini-review only new rat models of cortical lesions are discussed but we want to mention also that there is a new mouse model available on this topic20.
Different Types of Rat Models for Progressive MS Research
Figure 1 shows a scheme comparing the different models (a) - (d) side by side. It gives an overview of the different methods and highlights the distribution of the resulting cortical lesions.
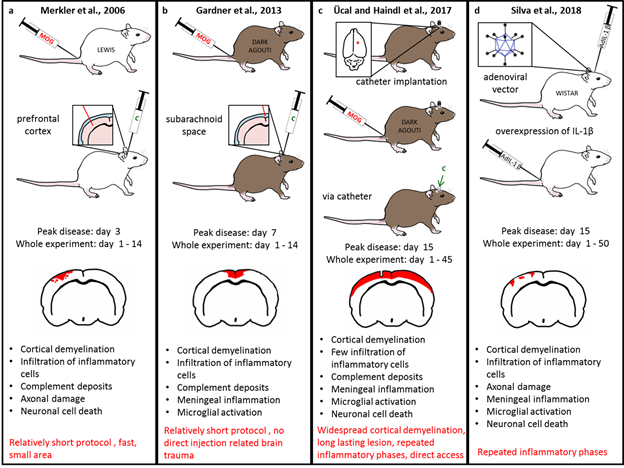
Figure 1: Schematic comparison of the four different animal models. In (a) the model of Merkler et al is represented. The protocol starts with the MOG immunization of Lewis rats. The proinflammatory cytokines are stereotactically injected directly into the prefrontal cortex indicated by the inlet beside the second rat schema. Peak disease is on day 3 and cortical demyelination is fully remyelinated until day 14. The model shows cellular features comparable with the cellular pathology of progressive MS, listed under the brain schema. The red spot in the brain schema indicates the lesion development which is only detectable on the ipsilateral hemisphere. The advantage are listed in red, it is a relatively short and fast protocol affecting a small cortex area but thereby reflecting good cellular comparability to human progressive MS. Panel (b) shows an overview of the animal model of Gardner et al. This model starts with the MOG immunization of Dark Agouti rats and cytokines are injected into the subarachnoid space. Peak disease of cortical demyelination is on day 7 and lesions are largely resolved until day 14. Cortical lesions appear mainly round the injection area affecting both hemispheres. Again most of the cellular features know for the progressive MS in humans are reflected in this model, listed under the brain schema. One big advantage (listed in red) is the avoidance of direct injection. Therefore, cellular reaction due to injection is very low. Panel (c) describes the animal model of Ücal and Haindl et al. This model starts with the implantation of a catheter directly into the cortex. After a healing period Dark Agouti rats are immunized with MOG and cytokines are given via the catheter. Peak disease is on day 15 and on day 30 some areas are already remyelinated. Via a second cytokine injection on day 30 another inflammatory phase could be induced lasting until day 45. The red rim in the brain schema indicates the long lasting widespread cortical demyelination. Again, the cellular features are listed under the brain schema and the advantages of the model are written in red. In (d) the animal model of Silva et al is shown. This is the only model discussed here, using adenoviral vectors for an overexpression of IL-1beta. The experiment starts with a central injection of AdIL-1beta into the cortex of Wistar rats. After 21 days a repeated inflammatory stimulus is given by peripheral injection of AdIL-1beta iv. Cortical demyelination peaked at day 15 which starts to resolve on day 30. Note, that the brain scheme only shows presumed lesion pattern since the authors don’t give an overview over the demyelinated hemisphere in their original work. Again, this model shows several cellular features known to be present in human progressive disease phase. The advantage of this model is given in red, also here repeated inflammatory phases are possible suitable for longer experimental approach.
Merkler et al., 2006: A new focal EAE model of cortical demyelination
In this work, lesions were stereotactically targeted to the cortex by injection of MOG-pre-immunized Lewis rats with pro-inflammatory cytokines (in particular a mixture of tumor necrosis factor-alpha (TNF-alpha) and interferon gamma (IFN-gamma)). The authors were able to generate highly reproducible demyelinated lesions in the neocortex with a close histological resemblance to cortical MS lesions. Their main methods focus on immunohistochemistry including quantification. They could observe the typical pattern of intracortical and subpial demyelination, infiltration with inflammatory cells, complement deposition, acute axonal damage and neuronal cell death. The cortical inflammation however largely resolved within two weeks along cortical demyelination, which was remyelinated within this time frame as well. They conclude that cortical inflammation is probably a transient phenomenon and with rapid remyelination15, even after multiple rounds of cortical demyelination induced by repetitive intracerebral injections21.
Gardner et al., 2013: Cortical grey matter demyelination can be induced by pro-inflammatory cytokines in the subarachnoid space of MOG-immunized rats
The authors injected pro-inflammatory cytokines (TNF-alpha and IFN-gamma) into the subarachnoid space of MOG-pre-immunized female Dark Agouti rats. Their main methods focus on immunohistochemistry including immunofluorescence and associated quantification. They could observe infiltration of lymphocytes into the meninges and extensive subpial demyelination. Also, extensive microglial activation was present from the pial surface to deeper cortical layers. The authors support the hypothesis that pro-inflammatory molecules produced in the meninges play a major role in cortical demyelination in MS and they also emphasize the involvement of an anti-myelin immune response17.
Ücal and Haindl et al., 2017: Widespread cortical demyelination can be induced by injection of pro-inflammatory cytokines via an implanted catheter in the cortex of MOG-immunized rats
This animal model is inspired by Merkler et al and Gardner et al with some modifications. It starts with the implantation of a tissue and MRI compatible catheter into the cortex of Dark Agouti rats and therefore offers direct access to the brain at any time without extra tissue trauma. After a healing period of two weeks, rats are immunized with a low dose of MOG. Four weeks later, with an IgG anti-MOG antibody established, animals receive an injection of pro-inflammatory cytokines (TNF-alpha and IFN-gamma) through the catheter directly into the cortex. Animals appeared only slightly fatigued between days 9-15 after cytokine injection, but no motor symptoms appeared. Further evaluation was done through immunohistochemistry and immunofluorescence. The authors observed demyelination of cortical and subcortical areas accompanied by remarkable microglial activation, neuronal loss, and astrocytic apoptosis. A second injection of cytokines four weeks after the first, led to a second demyelination phase with the same mild symptoms, but also to a striking global brain atrophy. Cytokine injection into the brain alone did not lead to cortical demyelination in this study, which was in line with earlier observations of both Merkler et al., 2006 and Gardner et al., 2013. The authors thus suggest that additional immunization against MOG or other myelin components is required to induce cortical demyelination.
Silva et al., 2018: A new focal model resembling features of cortical pathology of the progressive forms of MS
The authors developed an animal model based on innate immune response with an overexpression of interleukin 1 beta (IL-1beta) in the cortex of Wistar rats using an adenoviral vector (AdIL-1beta). They injected ADIL-1beta directly into the cortex and after 21 days also intravenously. Their main methods are behavioral tests and electron microscopy with corresponding quantifications. They could observe meningeal inflammation close to cortical lesions, demyelination, glial activation and neurodegeneration along with cognitive symptoms and MRI in their animals. With systemic pro-inflammatory stimulation (peripheral injection of ADIL-1beta) they could influence the cortical lesions and behavioral symptoms. They suggest that cognitive impairment could be affected by systemic inflammation in MS patients. They found that meningeal inflammation is associated with IL-1beta induced cortical lesions. Reflecting their results the authors suggest that meningeal inflammation should be considered a key factor in cortical lesions although its role in cortical pathology needs further research regarding meningeal inflammation induces cortical lesions or vice versa19.
Discussion
Aim of all animal models is a close resemblance of the human progressive MS pathology. The main features of the progressive disease phase are extensive cortical demyelination, diffuse axonal injury in the NAWM and meningeal inflammation. These features can be detected by histology and to a lesser extent also by MRI and behavioral testing. Immunohistochemistry is the most important method for tracking cellular changes during disease course and it is - when established properly - the most reliable one. While for MS patients MRI is most important, MRI results in animals have to be interpreted with caution due to methodical limitations22. An association between cortical lesions and cognitive impairment in MS patients has already been reported and for further research behavioral testing of rats in the course of the discussed experimental models is desired to improve knowledge in this field22.
All discussed animal models have their own focus and advantages. The work of Merkler et al., 2006 and Gardner et al., 2013 can be seen as pioneer work for the following models. They both show impressive results in a comparable fast protocol suitable for research of various cellular changes at distinct days. The model of Merkler et al. only the ipsilateral hemisphere was affected which offers the opportunity to work locally and predictably, when a focal pathology is required. In the model of Gardner et al. a larger area is affected with a comparable methodical effort offering some more cellular features like meningeal inflammation and microglia activation. However, when using these models one has to consider that the injection trauma per se will result in a cellular response.
The model of Ücal and Haindl et al., 2017 distinguishes from the others by the implanted catheter, offering an access to the cortex anytime for intracerebral injection while avoiding tissue injury accompanied by cellular interference. Comparable to Gardner et al. it offers a widespread cortical demyelination of both cortical hemispheres. In this model, the cortical pathology is even longer lasting (detectable even on day 30 after cytokine injection and beyond) and suitable for studying cellular mechanisms and chronicity.
In the models of Merkler et al., Gardner et al. as well as Ücal and Haindl et al. pro-inflammatory cytokines play a crucial role in augmenting the inflammatory response in the CNS15. TNF-alpha and IFN-gamma are also necessary for the induction of a second inflammatory response in the model of Ücal and Haindl et al., 2017.
The model of Silva et al., 2018 focuses on behavioral testing and correlation between cortical lesions and behavioral changes. Even though behavioral testing could be performed in any of the described animal models, the model of Silva et al., 2018 is the only model where pre-data already exists upon this topic. It has to be mentioned that models which offer the opportunity of repeated inflammatory phases also come up with higher effort.
Depending on the research question all models are suitable for MRI, histological investigation, behavioral testing and drug development. For research on small affected areas Merkler et al., 2006; Gardner et al., 2013 and Silva et al., 2018 would be the choice. If research on widespread and long lasting cortical demyelination of both hemispheres is desired Ücal and Haindl et al., 2017 should be preferred. For long-term experiments with repeated inflammatory phases the model of Ücal and Haindl et al., 2017 and Silva et al., 2018 is the right choice.
Taken together the reviewed rat models already contributed to the understanding of progressive MS pathology even though there is still need for improvement and further research. New findings of cortical lesions include the suggestion that cortical inflammation might be, at least at the beginning, a transient phenomenon and remyelination of cortical inflammatory demyelinating lesions may occur rapidly. Cortical demyelination seems to be influenced by pro-inflammatory molecules produced in the meninges and the requirement of an immunoreactivity against myelin components to induce cortical lesions. Meningeal inflammation is associated with IL-1beta induced cortical lesions and should be considered a key factor in cortical lesions development15,17–19.
Acknowledgement
A special thanks goes to all research groups working on new animal models researching the progressive MS pathology. Thanks for the effort and contribution to this topic.
References
- Gulani V, Calamante F, Shellock FG, et al. Gadolinium deposition in the brain: summary of evidence and recommendations. Lancet Neurol. 2017; 16: 564-570.
- Antel J, Antel S, Caramanos Z, et al. T. Primary progressive multiple sclerosis: Part of the MS disease spectrum or separate disease entity? Acta Neuropathol. 2012; 123: 627-638.
- Losy J. Is MS an inflammatory or primary degenerative disease? J. Neural Transm. 2013; 120: 1459-1462.
- Lassmann H. What drives disease in multiple sclerosis: Inflammation or neurodegeneration? Clin. Exp. Neuroimmunol. 2010; 1: 2-11.
- Zaratin P, Comi G, Leppert D. ‘Progressive MS – macro views’: The need for novel clinical trial paradigms to enable drug development for progressive MS. Mult Scler. 2017; 23: 1649-1655.
- Kutzelnigg A, Lucchinetti CF, Stadelmann C, et al. Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain. 2005; 128: 2705-2712.
- Peterson JW, Bö L, Mörk S,et al. Transected neurites, apoptotic neurons, and reduced inflammation in cortical multiple sclerosis lesions. Ann Neurol. 2001; 50: 389-400.
- Haider L, Zrzavy T, Hametner S, et al. The topograpy of demyelination and neurodegeneration in the multiple sclerosis brain. Brain. 2016; 139: 807-815.
- Magliozzi R, Howell O, Vora A, et al. Meningeal B-cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain. 2007; 130: 1089-1104.
- Choi SR, Howell OW, Carassiti D, et al. Meningeal inflammation plays a role in the pathology of primary progressive multiple sclerosis. Brain. 2012; 135: 2925-2937.
- Watzlawik J, Warrington AE, Rodriguez M. Importance of oligodendrocyte protection, BBB breakdown and inflammation for remyelination. Expert Rev Neurother. 2010; 10: 441-457.
- Gold R, Hartung HP, Toyka KV. Animal models for autoimmune demyelinating disorders of the nervous system. Mol Med Today. 2000; 6: 88-91.
- Basoglu H, Boylu NT, Kose H. Cuprizone induced demyelination in wistar rats: electrophysiological and histoligcal assesment. Euroean Rev. Med. Pharmacolical Sci. 2013; 20: 2711-2717.
- Tsunoda I, Fujinami RS. Neuropathogenesis of Theiler’s Murine Encephalomyelitis Virus Infection, An Animal Model for Multiple Sclerosis. J Neuroimmune Pharmacol. 2010; 5: 355-369.
- Merkler D, Ernsting T, Kerschensteiner M, et al. A new focal EAE model of cortical demyelination: Multiple sclerosis-like lesions with rapid resolution of inflammation and extensive remyelination. Brain. 2006; 129: 1972-1983.
- Pryce G, O'Neill JK, Croxford JL, et al. Autoimmune tolerance eliminates relapses but fails to halt progression in a model of multiple sclerosis. J. Neuroimmunol. 2005; 165: 41-52.
- Gardner C, Magliozzi R, Durrenberger PF, et al. Cortical grey matter demyelination can be induced by elevated pro-inflammatory cytokines in the subarachnoid space of MOG-immunized rats. Brain. 2013; 136: 3596-3608.
- Ücal M, Haindl MT, Adzemovic MZ, et al. Widespread cortical demyelination of both hemispheres can be induced by injection of pro-inflammatory cytokines via an implanted catheter in the cortex of MOG-immunized rats. Exp. Neurol. 2017; 294: 32-44.
- Silva BA, Leal MC, Farías MI, et al. A new focal model resembling features of cortical pathology of the progressive forms of multiple sclerosis: Influence of innate immunity. Brain Behav Immun. 2018; 69: 515-531.
- Lagumersindez-Denis N, Wrzos C, Mack M, et al. Differential contribution of immune effector mechanisms to cortical demyelination in multiple sclerosis. Acta Neuropathol. 2017; 134: 15-34.
- Rodriguez EG, Wegner C, Kreutzfeldt M, et al. Oligodendroglia in cortical multiple sclerosis lesions decrease with disease progression, but regenerate after repeated experimental demyelination. Acta Neuropathol. 2014; 128: 231-246.
- Nelson F, Datta S, Garcia N, et al. Intracortical Lesions by 3T Magnetic Resonance Imaging and Correlation with Cognitive Impairment in Multiple Sclerosis. 2009; 19: 389-399.