
A Case of Misdiagnosed Juvenile Dermatomyositis
Wang Liang1, Xu Min2, Sun Yiming3, Zhang Cheng1*
Abstract
Objective: To report a misdiagnosed case in order to avoid similar misdiagnosis of atypical juvenile dermatomyositis as muscular dystrophy in neurological clinics.
Methods: The process of misdiagnosis was first described. Thereafter, the patient’s diagnosis and treatment effect at the six-month follow-up were evaluated, and reported here.
Results: The patient presented an atypical course of juvenile dermatomyositis. The rashes, which mainly presented as erythema and wheals, were subsequently followed by the appearance of muscle weakness. However, the rashes were inconspicuous at first consult. The misdiagnosis of Duchenne muscular dystrophy was made due to the age of onset, distribution of muscle weakness, a high creatine kinase level, and other serum enzymatic changes. Upon further analysis, however, a diagnosis of juvenile dermatomyositis was definitively confirmed and the disease was controlled following systematic treatment for five months.
Conclusion: Although atypical juvenile dermatomyositis has a manifestation similar to that of muscular dystrophy, the considerations outlined in this case report will be helpful to avoid similar misdiagnosis.
Introduction
Dermatomyositis (DM), which is an idiopathic inflammatory myopathy (IIM), is a multisystem disorder that primarily affects skin and skeletal muscle1. Typical DM presents as either subacute, progressive weakness of proximal muscles; skin rashes; or both2. Typical lesions include periorbital heliotrope rashes, Gottron papules over extensor surfaces, and generalized photosensitive erythema3. Juvenile dermatomyositis (JDM) is a subtype of DM, where the median age of onset is 7 years4. A case report of a JDM case, which was misdiagnosed as urticarial vasculitis and Duchenne muscular dystrophy (DMD), by dermatologists and neurologists, respectively, is reported here. The reasons why this misdiagnosis occurred have also been analyzed. Further, some considerations that doctors need to take into account to avoid misdiagnosis of similar cases have also been raised.
Case history
A 3-year-old boy came to our outpatient department of neuromuscular disease, complaining of motor dysfunction, which included difficulty in climbing stairs, frequent falls when walking, and standing up from squatting slowly with the aid of hands placed on the ground, for several months. The muscle strength of the proximal upper and lower limbs was IV+ and IV-, respectively. The muscle strength of the distal upper limbs was V, while the distal lower limbs could not be evaluated due to poor compliance. The pseudo-hypertrophy of calves and abnormal gait were not obvious. Although a winged scapula was observed, there was no muscular tenderness. No skin lesions were observed during physical examination. There was nothing significant to note in the patient’s family history. Laboratory tests showed that the levels of AST, CK, CK-MB, and LDH were 280 u/L, 13,357 u/L, 814 u/L, and 1,637 u/L, respectively. The electrocardiograph and ultrasonic cardiogram were normal. Although there was no delay in the boy’s motor milestones, a first diagnosis of DMD was considered due to his manifestations and the CK elevation. A DMD gene test was conducted using multiplex ligation-dependent probe amplification (MLPA).
A month later, the test for copy number variation of the DMD gene showed a negative outcome. Since the patient’s mother, who was anxious about the diagnosis, had done her own search for detailed information about the disease, she conveyed the fact that her son had expressed a normal level of transaminase during a health examination that was conducted at kindergarten admittance. Since the transaminase level in DMD is always high5, this fact casted doubt on the DMD diagnosis.
A more detailed physical examination was conducted during the second consult. Some erythema and wheals on the forehead that had not appeared during the initial consult were found (Figure 1A). When asked, the patient’s mother informed us that the rashes had initially occurred one year prior, but had mostly disappeared. Erythematous rashes had first appeared at dorsal aspects of the metacarpophalangeal joints (Gottron sign), before extending to the forehead, eyelids and periorbit, neck, back, hips, legs, elbows, and knees. Some rashes were itchy at night and presented wheals when scratched (Figure 1C). The patient had consulted dermatologists several times before muscle weakness occurred. Autoantibodies were negative. According to the patient’s manifestations, most dermatologists made a diagnosis of urticarial vasculitis. Due to poor compliance, prednisone had been used for only a short time. The skin lesions, which had disappeared gradually, even in the absence of systematic treatment, were mild at the initial consult.
Therefore, JDM was considered as the first diagnosis. Although lacking electromyography or muscle biopsy, the evidence was sufficient to diagnose probable DM according to Bohan and Peter’s diagnostic criteria2. Invasive procedures like electromyography and muscle biopsy were temporarily halted6, and the patient was treated using oral prednisone, intravenous steroid pulse therapy, and intravenous immunoglobulin pulse therapy. The rashes disappeared and motor function clearly improved following a 5-month duration of treatment (Figure 1B, D). Further, the level of CK had decreased to 800 u/L. Thus, the JDM diagnosis could be confirmed.
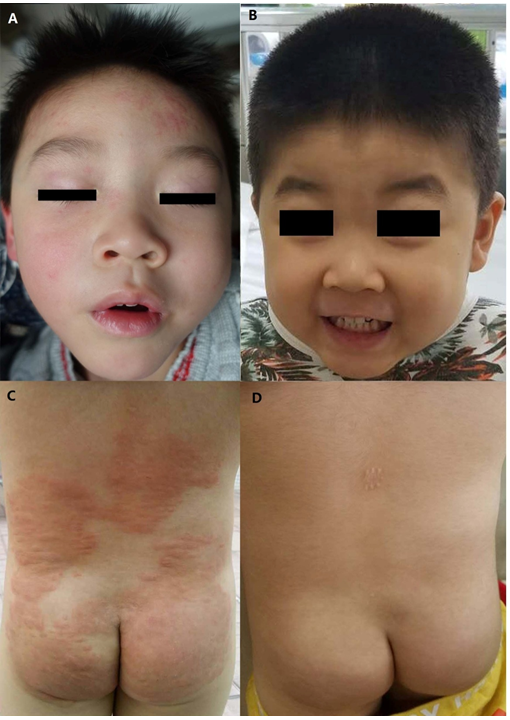
Figure 1: Dermatological symptoms presented by the patient and their resolution following treatment.
(A) The rashes that were present on the patient’s forehead during the second consult. (B) Face without rashes after regular treatment for five months. (C) Wheals on the back and hips, prior to the onset of muscle weakness. (D) Back and hips without wheals after regular treatment for five months.
Discussion
DMD is a common hereditary neuromuscular disease, which has an age of onset during childhood and an incidence of 1 in 3600–6000 live male newborns. In contrast, JDM has a relatively low incidence of 3.2 in 1, 000, 0003,7. Atypical JDM, such as the case presented here, is rarer. The main characteristic the patient presented with during the initial consult was muscle weakness for several months, without muscle tenderness. There were no observable rashes during the physical examination. Further, the patient’s level of CK was elevated highly and autoantibodies were negative. Given the higher incidence of DMD, this was considered as the first diagnosis, although JDM was not excluded. During the second consult, given new information about past conditions of transaminase level and rashes, the JDM diagnosis was re-considered. In fact, this misdiagnosis could have been avoided if these points had been highlighted during the first consult.
Therefore, three key points led to misdiagnosis in the present case. Firstly, the level of transaminase before onset of disease was not clear during the first consult, and we did not ask about related conditions. Secondly, the rashes were not obvious during the initial physical examination. Finally, although typical rashes of DM include periorbital heliotrope rashes and Gottron papules, in the current case, wheals were very heavy and covered up the typical rashes, and had existed alone for about half a year before muscle weakness occurred, which led to misdiagnosis as ‘urticarial vasculitis’ by dermatologists. Thus, the patient’s mother believed that the rashes were not related to muscle weakness, and this symptom was ignored by both the patient’s mother and the neurologists. If any of the points above had been noticed at the initial consult, this misdiagnosis would be unlikely to occur.
Conclusion
DMD and some JDM have similar muscle weakness and changes in serum enzymes, such as ALT, AST, and CK. As genetic testing has become more common, invasive muscle biopsy, which can easily differentiate muscular dystrophy from IIM, is not the first or the primary means considered to make a definite diagnosis. Therefore, when considering a diagnosis of DMD, neurologists should also consider JDM and check the patient’s previous history of serum indexes, especially ALT, AST, and CK levels. Further, it is also necessary to investigate the presence of rashes, even if these are not evident during physical examination. These two considerations would be helpful to make a correct diagnosis of JDM.
Acknowledgement
This study was supported by grants from Natural Science Foundation of China (81471280 & 81271401), Guangdong Province Science and Technology Plan (2014A020212130).
References
- Santmyire-Rosenberger B, Dugan EM. Skin involvement in dermatomyositis. Curr Opin Rheumatol. 2003; 15: 714-722.
- Findlay AR, Goyal NA, Mozaffar T. An overview of polymyositis and dermatomyositis. Muscle Nerve. 2015; 51: 638-656.
- Robinson AB, Reed AM. Clinical features, pathogenesis and treatment of juvenile and adult dermatomyositis. Nat Rev Rheumatol. 2011; 7: 664-675.
- Lowry CA, Pilkington CA. Juvenile dermatomyositis: extramuscular manifestations and their management. Curr Opin Rheumatol. 2009; 21: 575-580.
- McMillan HJ, Gregas M, Darras BT, et al. Serum transaminase levels in boys with Duchenne and Becker muscular dystrophy. Pediatrics. 2011; 127: e132-136.
- Ramanan AV, Feldman BM. Clinical outcomes in juvenile dermatomyositis. Curr Opin Rheumatol. 2002; 14: 658-662.
- Bushby K, Finkel R, Birnkrant DJ, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010; 9: 77-93.