
The Rag GTPases-mTORC1-TFE3 Axis in Kidney Cancer and Neurodevelopment: A Mini Review
Christopher Nardone1,2, Xin Gu3, Stephen J. Elledge1,2*
1Division of Genetics, Department of Medicine, Howard Hughes Medical Institute, Brigham and Women’s Hospital, Boston, MA, USA
2Department of Genetics, Harvard Medical School, Boston, MA, USA
3Department of Neurobiology, Harvard Medical School, Boston, MA, USA
Abstract
The MiT/TFE proteins (MITF, TFE3, TFEB, and TFEC) constitute a family of transcription factors that maintain cellular homeostasis by controlling the transcription of genes involved in lysosomal biogenesis, autophagy, oxidative metabolism, and pigmentation. While significant effort has been placed in understanding the downstream function of this family, the upstream regulation has only recently become clearer. It was appreciated that the nutrient-sensing Rag GTPases-mTORC1 pathway attenuates the activity of this family, but how this occurs mechanistically remained unclear. The physiological role of the MiT/TFE family is underscored by the fact that numerous human diseases are caused by their misregulation. The goal of this minireview is to provide a summary of recent findings that have elucidated how this family is regulated upstream and, hence, the molecular basis of a rare kidney cancer and neurodevelopmental syndrome caused by mutations in TFE3. The information presented here may serve as a framework for future studies.
Summary of the nutrient-sensing Rag GTPases-mTORC1 pathway
Organisms must sense dietary fluctuations to appropriately adapt to a changing environment. When nutrients such as amino acids are replete, organisms promote anabolism and suppress catabolism. At a cellular and molecular level, this is achieved by a master regulator called mTORC1, for mechanistic target of rapamycin complex 11,2. When active, this protein kinase complex phosphorylates many downstream targets, leading to increased protein and nucleotide synthesis, and decreased autophagy. The activity of mTORC1 must be tightly controlled so the kinase remains active when nutrients are available to support anabolism but inactive when nutrients are scarce. mTORC1 is directly recruited to the lysosomal surface by the Rag GTPases that are anchored to the lysosomal surface by the heteropentameric Ragulator complex3-5. The Rag proteins function as obligate heterodimers in which the active complex consists of RagA or B bound to GTP, and Rag C or D bound to GDP. Once recruited, the mTORC1 kinase is directly activated by another lysosome tethered GTPase called Rheb when bound to GTP6-10. The presence of amino acids and growth factors respectively maintain the Rags proteins and Rheb in the active state, thereby activating mTORC1. However, dedicated GTPase-activating proteins (GAPs) are turned on during starvation to inactivate mTORC1. For example, during amino acid deprivation, the GATOR1 complex negatively regulates mTORC1 signaling by stimulating the GTPase activity of RagA/B, leading to mTORC1 cytosolic redistribution11-13. In contrast, TSC (Tuberous sclerosis complex) negatively regulates mTORC1 signaling in the absence of growth factors by stimulating the GTPase activity of Rheb6-10. Thus, organisms evolved an elegant system centered on lysosome tethered GTPases that integrate nutrient and growth factor availability to control the master metabolic kinase, mTORC1.
Overview of the MiT/TFE transcription factors
The purpose of this minireview is to highlight recent findings related to a small family of transcription factors that lie directly downstream of the Rag GTPases-mTORC1 pathway. The MiT/TFE proteins (MITF, TFE3, TFEB, and TFEC) constitute a family of basic helix-loop-helix leucine zipper (bHLH-ZIP)-containing proteins that function as homo- or hetero-dimers to maintain cellular homeostasis14,15. TFE3 and TFEB are master regulators of lysosomal biogenesis, promote autophagy flux, lipid oxidation, and are involved in a plethora of other processes16-19. This is achieved in part by binding a modified E-box DNA motif called the CLEAR element (Coordinated Lysosomal Expression And Regulation) that is enriched in the promoter region of lysosomal membrane proteins, lysosomal hydrolases, and autophagy adaptors16. In contrast, MITF is a master regulator of the melanocytic lineage and regulates melanin production by binding an M-box DNA sequence motif enriched in the promoter region of pigmentation genes20,21. Much less is known about the biological functions of TFEC. These transcription factors are generally catabolic in nature; therefore, it is important for cells to limit their activity in nutrient replete conditions.
Upstream regulation of the MiT/TFE family by the Rag GTPases-mTORC1 pathway
In the presence of nutrients, MITF, TFE3, and TFEB are heavily phosphorylated by mTORC1 to inhibit transcriptional activity22-25. However, unlike canonical mTORC1 substrates including 4EBP1 and S6K1, these transcription factors are unique because they require recruitment to the lysosomal surface where mTORC1 is localized for phosphorylation to occur. Current literature posited that active Rag GTPases recruit the MiT/TFE family to the lysosomal surface, leading to phosphorylation by mTORC1 on a conserved serine residue to create a binding site for the 14-3-3 chaperone proteins that sequester the transcription factors in the cytosol26. How these transcription factors are recruited by the Rag GTPases and whether additional layers of regulation exist to control transcriptional activity remained unclear until recently.
We found that apart from the 14-3-3 site, an additional phosphosite is present in TFE3 and MITF, but not TFEB, to suppress transcriptional activity by promoting TFE3 and MITF protein degradation27. Specifically, we show that TFE3 and MITF are recruited to the lysosomal surface by the Rag GTPases, leading to phosphorylation of a conserved serine residue (Figure 1). This creates a binding site, called a degron, that is captured by the CUL1b-TrCP ubiquitin ligase complex for ubiquitination and subsequent proteasomal degradation. In the absence of nutrients such as amino acids, TFE3 and MITF are not recruited by the Rag GTPases and are hypo-phosphorylated, leading to marked stabilization, nuclear translocation, and transcription of downstream genes. This phospho-degron plays a central role in the control of TFE3 and MITF by nutrients because de-phosphorylation of the degron serine is necessary and partially sufficient to activate TFE3 and MITF. While the CUL1b-TrCP and 14-3-3 regulation now appear to be the key layers regulating TFE3 and MITF activity, there may exist additional phosphosites in TFE3 and MITF that could contribute to inactivation. A systematic interrogation of TFE3 and MITF phosphosites on transcriptional activity is needed to further understand whether other layers of regulation may exist. To do this, the Rag GTPases, mTORC1, and MiT/TFE transcription factors should be reconstituted in vitro from purified proteins and phosphorylation monitored by liquid chromatography-mass spectrometry. Candidate phosphosites should be tested for their impact on transcriptional activity in cells by complementing the phosphosite mutants in MiT/TFE knockout cells.
Recurrent loss of the CUL1b-TrCP degron in oncogenic TFE3 genomic translocations
The physiological importance of the MiT/TFE family is underscored by the fact that several human diseases are caused by their misregulation. For an overview of the diseases not discussed in this minireview, please see this excellent recent review article28. We focus here on two divergent pathologies, a rare kidney cancer and neurodevelopmental syndrome, that are caused by mutations in TFE3. The TFE3 gene is located on the X chromosome and in rare cases, a double strand DNA break occurs within fragile sites of TFE3 (less commonly in TFEB and MITF) that results in fusion of the broken chromosome with other regions of the genome29-31. This translocation and fusion event lead to the production of a novel protein, where the N-terminus of diverse partners fuses with the C-terminal DNA-binding domain of TFE3. These chimeric proteins cause translocation renal cell carcinoma (tRCC), a rare cancer with an incidence of 1-5% in adult, and more than 50% of all kidney cancers in children29. While the cellular origin of this disease remains unclear, we found that the N-terminal phospho-degron region in TFE3 is lost during the translocation event in almost all clinically reported tumors27. Thus, these oncogenic TFE3-fusion proteins are incredibly stable and escape proteasomal degradation. Modeling tRCC at an organismal level is warranted to understand the contribution of protein stability to oncogenicity, the role of the diverse partner genes, and whether a rare cell type exists in the kidney that is transformed by TFE3-fusion proteins, thereby driving the disease. It is possible that single cell RNA sequencing from biopsies of patient-derived tumors could be useful for identifying the putative transformed cell type(s).
TFE3 missense mutations in a neurodevelopmental syndrome
Beyond cancer, de novo germline missense mutations in TFE3 were recently described within a cohort of people diagnosed with a severe neurodevelopmental syndrome32-34. These people suffer from intellectual disability, speech impairment, an inability to walk, facial and skeletal abnormalities, metabolic imbalance including obesity, and regional skin hyperpigmentation. The missense mutations cluster in two adjacent regions of TFE3 that are highly conserved between MiT/TFE family members and fold into two alpha helices. We showed that these adjacent alpha helices are sufficient to confer an interaction with the Rag GTPases27. Specifically, the second helix forms an extensive interaction interface with a region of RagA and most of the disease associated residues are at the heart of this RagA-TFE3 interface. The remaining mutations that are found in the first helix of TFE3 can be explained by a beautiful structure of TFEB bound to the Rag GTPases and the disease associated residues are in a region that is crucial for interacting with RagC35. Therefore, these de novo gain-of-function missense mutations found in a neurodevelopmental syndrome prevent TFE3 recruitment to the Rag GTPases for phosphorylation by mTORC1, leading to a constitutively stable and transcriptionally active protein. While the molecular and structural basis for this disorder is now more clearly understood, the role of constitutively active TFE3 during development remains undefined. It was suggested that active TFE3 impairs the differentiation of mouse embryonic stem cells to neurons32. However, the consequence of TFE3 activation during development in a model organism is not established and the result of a constitutively active TFE3 in a developed nervous system is unknown. Future work should focus on modeling the TFE3 neurodevelopmental disorder in vivo.
Targeting mutant TFE3 as a potential therapy
Targeting transcription factors for therapies is particularly challenging due to a lack of druggable pockets naturally found in many enzymes36. Anti-sense oligonucleotides have emerged as a promising avenue to target undruggable proteins by harnessing the power of RNA interference and could be applied to oncogenic or constitutively active TFE337. Another potential route for targeting the mutant TFE3 that causes the neurodevelopmental syndrome is to develop small molecules that restore mutant TFE3 ubiquitination by the CUL1b-TrCP complex. The pathogenic missense mutations that occur in the Rag binding region of TFE3 lead to hypo-phosphorylation of the b-TrCP degron and a significant reduction in the binding affinity between TFE3 and the CUL1b-TrCP complex. Molecular glue degraders are small molecules that potently increase the binding affinity between weak ubiquitin ligase-substrate interactions, thereby facilitating the degradation of the substrate38,39. A screen to identify small molecules that restore a high affinity interaction between mutant TFE3 and b-TrCP could be an attractive option. This has been accomplished for mutant b-catenin with b-TrCP, and thus the uncovered molecules could be repurposed or redesigned to restore the mutant TFE3-b-TrCP interaction40.
Neurological disorders associated with elevated mTORC1 signaling
Finally, we would like to conclude this minireview by highlighting the fact that elevated mTORC1 signaling has been associated with numerous neurodevelopmental and neuropsychiatric disorders41,42 (Figure 1). Heterozygous germline loss-of-function mutations in TSC lead to hyperactive mTORC1, causing cortical development malformation and disfunction of neural circuits, which contribute directly to severe epilepsy and cognitive deficits43. A somatic gain-of-function mutant in Rheb is associated with focal cortical dysplasia and in mice this Rheb mutant causes seizures44. Heterozygous germline loss-of-function mutations in components of the GATOR1 or KICSTOR complex, which localizes GATOR1 to the lysosomal surface thus also serving as a negative regulator of mTORC145, were reported to cause familial focal epilepsy and focal cortical dysplasia46-48. Gain-of-function mutations in mTOR itself cause Smith-Kingsmore syndrome, which is characterized by intellectual disability, macrocephaly, seizures, autism spectrum, and attention-deficit/hyperactivity disorders49. A central theme to the disorders highlighted in Figure 1 is that mTORC1 signaling is elevated. Interestingly, it was shown that active TFE3 and TFEB strongly enhance mTORC1 signaling by directly promoting the transcription of RagD50. Perhaps the gain-of-function missense mutations in TFE3 that cause a neurodevelopmental syndrome may also be explained by elevated mTORC1 signaling. It remains unclear how hyperactive mTORC1 causes defects in neuronal development. Future research should focus on investigating the cellular origin of these mTORC1-related neurological diseases using animal models, whether similar type(s) of brain cells are involved across diverse mutations that cause diseases, and explore the possibility of harnessing gene therapy, anti-sense oligonucleotides, or small molecules such as rapamycin to treat the symptoms.
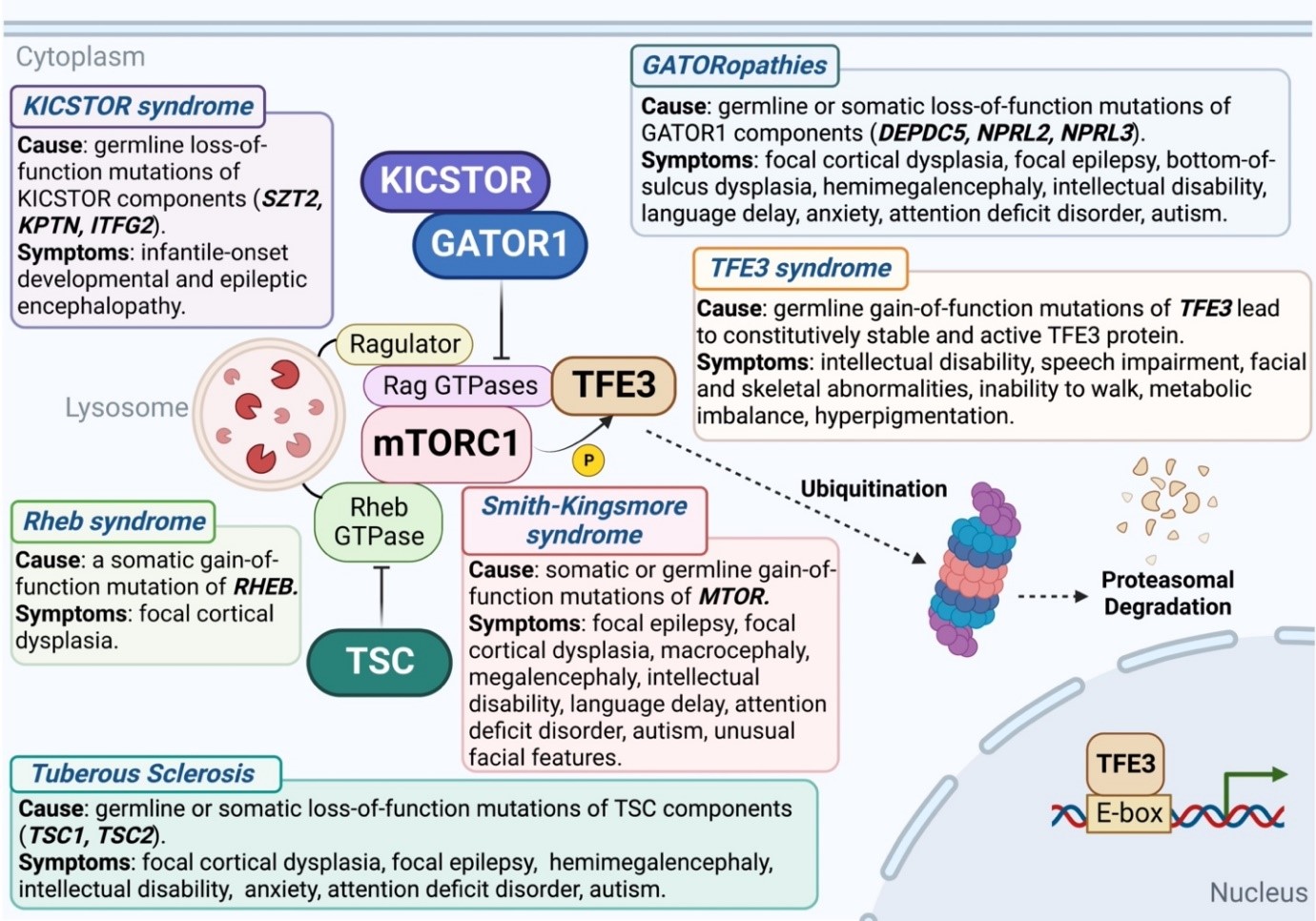
Figure 1: Mutations in various components of the mTORC1 pathway that lead to elevated signaling cause neurodevelopmental and neuropsychiatric disorders. Figure generated using BioRender.
Acknowledgements
We apologize in advance to our colleagues whose work could not be cited owing to space limitations. C.N. is supported by the National Science Foundation Graduate Research Fellowship Program. X.G. is the National Mah Jongg League Fellow of the Damon Runyon Cancer Research Foundation (DRG-2469-22). S.J.E. is an investigator with the Howard Hughes Medical Institute.
Conflict of Interests
S.J.E. is a founder of TSCAN Therapeutics, MAZE Therapeutics, ImmuneID, and Mirimus, serves on the scientific advisory boards of Homology Medicines, ImmuneID, MAZE Therapeutics, X- Chem, and TSCAN Therapeutics, and is an advisor for MPM Capital. Other authors declare no competing interests.
References
- Saxton RA, Sabatini DM. mTOR Signaling in Growth, Metabolism, and Disease. Cell. 2017; 168(6): 960-976.
- Liu GY, Sabatini DM. mTOR at the nexus of nutrition, growth, ageing and disease. Nat Rev Mol Cell Biol. 2020; 21(4): 183-203.
- Sancak Y, Peterson TR, Shaul YD, et al. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science. 2008; 320(5882): 1496-501.
- Kim E, Goraksha-Hicks P, Li L, et al. Regulation of TORC1 by Rag GTPases in nutrient response. Nat Cell Biol. 2008; 10(8): 935-45.
- Sancak Y, Bar-Peled L, Zoncu R, et al. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell. 2010; 141(2): 290-303.
- Inoki K, Li Y, Xu T, et al. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003; 17(15): 1829-34.
- Li Y, Inoki K, Guan KL. Biochemical and functional characterizations of small GTPase Rheb and TSC2 GAP activity. Mol Cell Biol. 2004; 24(18): 7965-75.
- Saito K, Araki Y, Kontani K, et al. Novel role of the small GTPase Rheb: its implication in endocytic pathway independent of the activation of mammalian target of rapamycin. J Biochem. 2005; 137(3): 423-30.
- Saucedo LJ, Gao X, Chiarelli DA, et al. Rheb promotes cell growth as a component of the insulin/TOR signalling network. Nat Cell Biol. 2003; 5(6): 566-71.
- Stocker H, Radimerski T, Schindelholz B, et al. Rheb is an essential regulator of S6K in controlling cell growth in Drosophila. Nat Cell Biol. 2003; 5(6): 559-65.
- Panchaud N, Péli-Gulli MP, De Virgilio C. Amino acid deprivation inhibits TORC1 through a GTPase-activating protein complex for the Rag family GTPase Gtr1. Sci Signal. 2013.
- Bar-Peled L, Chantranupong L, Cherniack AD, et al. A Tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science. 2013; 340(6136): 1100-6.
- Shen K, Huang RK, Brignole EJ, et al. Architecture of the human GATOR1 and GATOR1-Rag GTPases complexes. Nature. 2018; 556(7699): 64-69.
- Hemesath TJ, Steingrímsson E, McGill G, et al. microphthalmia, a critical factor in melanocyte development, defines a discrete transcription factor family. Genes Dev. 1994; 8(22): 2770-80.
- La Spina M, Contreras PS, Rissone A, et al. MiT/TFE Family of Transcription Factors: An Evolutionary Perspective. Front Cell Dev Biol. 2021; 8: 609683.
- Sardiello M, Palmieri M, di Ronza A, et al. A gene network regulating lysosomal biogenesis and function. Science. 2009; 325(5939): 473-7.
- Settembre C, Di Malta C, Polito VA, et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011; 332(6036): 1429-33.
- Martina JA, Diab HI, Lishu L, et al. The nutrient-responsive transcription factor TFE3 promotes autophagy, lysosomal biogenesis, and clearance of cellular debris. Sci Signal. 2014.
- Settembre C, De Cegli R, Mansueto G, et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat Cell Biol. 2013; 15(6): 647-58.
- Goding CR, Arnheiter H. MITF-the first 25 years. Genes Dev. 2019; 33(15-16): 983-1007.
- Kawakami A, Fisher DE. The master role of microphthalmia-associated transcription factor in melanocyte and melanoma biology. Lab Invest. 2017; 97(6): 649-656.
- Roczniak-Ferguson A, Petit CS, Froehlich F, et al. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci Signal. 2012.
- Settembre C, Zoncu R, Medina DL, et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012; 31(5): 1095-108.
- Martina JA, Puertollano R. Rag GTPases mediate amino acid-dependent recruitment of TFEB and MITF to lysosomes. J Cell Biol. 2013; 200(4): 475-91.
- Martina JA, Chen Y, Gucek M, et al. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy. 2012; 8(6): 903-14.
- Puertollano R, Ferguson SM, Brugarolas J, et al. The complex relationship between TFEB transcription factor phosphorylation and subcellular localization. EMBO J. 2018.
- Nardone C, Palanski BA, Scott DC, et al. A central role for regulated protein stability in the control of TFE3 and MITF by nutrients. Mol Cell. 2023; 83(1): 57-73.e9.
- Napolitano G, Di Malta C, Ballabio A. Non-canonical mTORC1 signaling at the lysosome. Trends Cell Biol. 2022; 32(11): 920-931.
- Kauffman EC, Ricketts CJ, Rais-Bahrami S, et al. Molecular genetics and cellular features of TFE3 and TFEB fusion kidney cancers. Nat Rev Urol. 2014; 11(8): 465-75.
- Perera RM, Di Malta C, Ballabio A. MiT/TFE Family of Transcription Factors, Lysosomes, and Cancer. Annu Rev Cancer Biol. 2019; 3: 203-222.
- Bakouny Z, Sadagopan A, Ravi P, et al. Integrative clinical and molecular characterization of translocation renal cell carcinoma. Cell Rep. 2022; 38(1): 110190.
- Villegas F, Lehalle D, Mayer D, et al. Lysosomal Signaling Licenses Embryonic Stem Cell Differentiation via Inactivation of Tfe3. Cell Stem Cell. 2019; 24(2): 257-270.e8.
- Lehalle D, Vabres P, Sorlin A, et al. De novo mutations in the X-linked TFE3 gene cause intellectual disability with pigmentary mosaicism and storage disorder-like features. J Med Genet. 2020; 57(12): 808-819.
- Diaz J, Berger S, Leon E. TFE3-associated neurodevelopmental disorder: A distinct recognizable syndrome. Am J Med Genet A. 2020; 182(3): 584-590.
- Cui Z, Napolitano G, de Araujo MEG, et al. Structure of the lysosomal mTORC1-TFEB-Rag-Ragulator megacomplex. Nature. 2023; 614(7948): 572-579.
- Henley MJ, Koehler AN. Advances in targeting 'undruggable' transcription factors with small molecules. Nat Rev Drug Discov. 2021; 20(9): 669-688.
- Rinaldi C, Wood MJA. Antisense oligonucleotides: the next frontier for treatment of neurological disorders. Nat Rev Neurol. 2018; 14(1): 9-21.
- Cao S, Kang S, Mao H, et al. Defining molecular glues with a dual-nanobody cannabidiol sensor. Nat Commun. 2022; 13(1): 815.
- Schreiber SL. The Rise of Molecular Glues. Cell. 2021; 184(1): 3-9.
- Simonetta KR, Taygerly J, Boyle K, et al. Prospective discovery of small molecule enhancers of an E3 ligase-substrate interaction. Nat Commun. 2019; 10(1): 1402.
- Ryskalin L, Limanaqi F, Frati A, et al. mTOR-Related Brain Dysfunctions in Neuropsychiatric Disorders. Int J Mol Sci. 2018; 19(8): 2226.
- Moloney PB, Cavalleri GL, Delanty N. Epilepsy in the mTORopathies: opportunities for precision medicine. Brain Commun. 2021; 3(4): fcab222.
- Henske EP, Jóźwiak S, Kingswood JC, et al. Tuberous sclerosis complex. Nat Rev Dis Primers. 2016; 2: 16035.
- Zhao S, Li Z, Zhang M, et al. A brain somatic RHEB doublet mutation causes focal cortical dysplasia type II. Exp Mol Med. 2019; 51(7): 1-11.
- Wolfson RL, Chantranupong L, Wyant GA, et al. KICSTOR recruits GATOR1 to the lysosome and is necessary for nutrients to regulate mTORC1. Nature. 2017; 543(7645): 438-442.
- Ricos MG, Hodgson BL, Pippucci T, et al. Mutations in the mammalian target of rapamycin pathway regulators NPRL2 and NPRL3 cause focal epilepsy. Ann Neurol. 2016; 79(1): 120-31.
- Baldassari S, Picard F, Verbeek NE, et al. The landscape of epilepsy-related GATOR1 variants. Genet Med. 2019; 21(2): 398-408.
- Iffland PH 2nd, Carson V, Bordey A, et al. GATORopathies: The role of amino acid regulatory gene mutations in epilepsy and cortical malformations. Epilepsia. 2019; 60(11): 2163-2173.
- Gordo G, Tenorio J, Arias P, et al. mTOR mutations in Smith-Kingsmore syndrome: Four additional patients and a review. Clin Genet. 2018; 93(4): 762-775.
- Di Malta C, Siciliano D, Calcagni A, et al. Transcriptional activation of RagD GTPase controls mTORC1 and promotes cancer growth. Science. 2017; 356(6343): 1188-1192.